usage: flye (--pacbio-raw | --pacbio-corr | --pacbio-hifi | --nano-raw |
--nano-corr | --nano-hq ) file1 [file_2 ...]
--out-dir PATH
[--genome-size SIZE] [--threads int] [--iterations int]
[--meta] [--polish-target] [--min-overlap SIZE]
[--keep-haplotypes] [--debug] [--version] [--help]
[--scaffold] [--resume] [--resume-from] [--stop-after]
[--read-error float] [--extra-params]
Assembly of long reads with repeat graphs
optional arguments:
-h, --help show this help message and exit
--pacbio-raw path [path ...]
PacBio regular CLR reads (<20% error)
--pacbio-corr path [path ...]
PacBio reads that were corrected with other methods (<3% error)
--pacbio-hifi path [path ...]
PacBio HiFi reads (<1% error)
--nano-raw path [path ...]
ONT regular reads, pre-Guppy5 (<20% error)
--nano-corr path [path ...]
ONT reads that were corrected with other methods (<3% error)
--nano-hq path [path ...]
ONT high-quality reads: Guppy5+ or Q20 (<5% error)
--subassemblies path [path ...]
[deprecated] high-quality contigs input
-g size, --genome-size size
estimated genome size (for example, 5m or 2.6g)
-o path, --out-dir path
Output directory
-t int, --threads int
number of parallel threads [1]
-i int, --iterations int
number of polishing iterations [1]
-m int, --min-overlap int
minimum overlap between reads [auto]
--asm-coverage int reduced coverage for initial disjointig assembly [not set]
--hifi-error float [deprecated] same as --read-error
--read-error float adjust parameters for given read error rate (as fraction e.g. 0.03)
--extra-params extra_params
extra configuration parameters list (comma-separated)
--plasmids unused (retained for backward compatibility)
--meta metagenome / uneven coverage mode
--keep-haplotypes do not collapse alternative haplotypes
--scaffold enable scaffolding using graph [disabled by default]
--trestle [deprecated] enable Trestle [disabled by default]
--polish-target path run polisher on the target sequence
--resume resume from the last completed stage
--resume-from stage_name
resume from a custom stage
--stop-after stage_name
stop after the specified stage completed
--debug enable debug output
-v, --version show program's version number and exitInput reads can be in FASTA or FASTQ format, uncompressed or compressed with gz. Currently, PacBio (CLR, HiFi, corrected) and ONT reads (regular, HQ, corrected) are supported. Expected error rates are <20% for PB CLR/regular ONT, <5% for ONT HQ, <3% for corrected, and <1% for HiFi. Note that Flye was primarily developed to run on uncorrected reads. You may specify multiple files with reads (separated by spaces). Mixing different read types is not yet supported. The --meta option enables the mode for metagenome/uneven coverage assembly.
To reduce memory consumption for large genome assemblies, you can use a subset of the longest reads for initial disjointig assembly by specifying --asm-coverage and --genome-size options. Typically, 40x coverage is enough to produce good disjointigs.
You can run Flye polisher as a standalone tool using --polish-target option.
You can try Flye assembly on these ready-to-use datasets:
The original dataset is available at the PacBio website. We coverted the raw bas.h5 file to the FASTA format for the convenience.
wget https://zenodo.org/record/1172816/files/E.coli_PacBio_40x.fasta
flye --pacbio-raw E.coli_PacBio_40x.fasta --out-dir out_pacbio --threads 4with the threads argument being optional (you may adjust it for your environment), and out_pacbio being the directory where the assembly results will be placed.
The dataset was originally released by the Loman lab.
wget https://zenodo.org/record/1172816/files/Loman_E.coli_MAP006-1_2D_50x.fasta
flye --nano-raw Loman_E.coli_MAP006-1_2D_50x.fasta --out-dir out_nano --threads 4The default mode for regular ONT data is --nano-raw. It works well for a good range of datasets, from old R7 pores to the most recent R9.x and R10.x. The expected error rate is 10-15%.
For the most recent ONT data basecalled with Guppy5 use the new --nano-hq mode. Expected error rate is <5%.
For Q20 data, use a combination of --nano-hq and --read-error 0.03.
If you have error-corrected ONT reads (with methods such as Canu), use --nano-corr.
The default mode for regular PacBio CLR data is --pacbio-raw. Works for a wide range of datasets (P5C3/P6C4/Sequel) with error rate 13-15%.
Note that in CLR mode Flye assumes that the input files represent PacBio subreads, e.g. adaptors and scraps are removed and multiple passes of the same insertion sequence are separated. This is typically handled by PacBio instruments/toolchains, however we saw examples of problemmatic raw -> fastq conversions, which resulted into incorrect subreads. In this case, consider using pbclip to fix your Fasta/q reads.
For PacBio HiFi use the --pacbio-hifi mode. The default error-rate is 0.001 (in HPC space), and works well for the default CCS algorithm settings (e.g. 3+ polymerase passes). Error could be adjusted via --read-error.
If you have error-corrected PacBio reads (with methods such as Canu), use --pacbio-corr.
WARNING: this mode is being deprecated and will be removed in the future versions. This is to make the future maintenance of Flye easier. Instead, we suggest to use more specialized software, like quickmerge.
--subassemblies input mode generates a consensus of multiple high quality contig assemblies (such as produced by different short/long read assemblers). The expected error rate is <1%. You might want to skip the polishing stage with --iterations 0 argument (however, it might still be helpful to correct small structural errors).
Flye works directly with base-called raw reads and does not require any prior error correction or trimming. Flye automatically detects chimeric reads or reads with low quality ends.
No longer required as input. However, it must be used in conjunction with --asm-coverage option.
This sets a minimum overlap length for two reads to be considered overlapping. In the latest Flye versions, this parameter is chosen automatically based on the read length distribution (reads N90) and does not require manual setting. Typical value is 3k-10k (and down to 1k for datasets with shorter read length). Intuitively, we want to set this parameter as high as possible, so the repeat graph is less tangled. However, higher values might lead to assembly gaps.
In some rare cases it makes sense to manually increase minimum overlap for assemblies of big genomes with long reads and high coverage.
Metagenome assembly mode. The main differences are that “regular” mode assumes a relatively uniform coverage of the assembled genome and makes certain desicions based on that. The metagenome mode is more general in this respect, and works well for assembly of complex microbial communities with highly non-uniform coverage and richer repeat content. It is sensitive to very short sequences and underrepresented organisms at low read coverage (as low as 3x).
For relatively complex single genomes, “regular” mode often outperforms metageomic mode.
By default, Flye (and metaFlye) collapses graph structures caused by alternative haplotypes (bubbles, superbubbles, roundabouts) to produce longer consensus contigs. The option --keep-haplotypes retains the alternative paths on the graph, producing less contigouos, but more detailed assembly.
Starting from the version 2.9 Flye does not perform scaffolding by default, which guarantees that all assembled sequences do not have any gaps. Scaffolding could still be enabled by adding --scaffold.
WARNING: this mode is being deprecated and will be removed in the future versions. This is to make the future maintenance of Flye easier.
Trestle is an extra module that resolves simple repeats of multipicity 2 that were not bridged by reads. Depending on the datasets, it might resolve a few extra repeats, which is helpful for small (bacterial genomes). Use --trestle option to enable the module. On large genomes, the contiguity improvements are usually minimal, but the computation might take a lot of time.
Typically, assemblies of large genomes at high coverage require several hundreds of RAM. For high coverage datasets, you can reduce memory usage by using only a subset of longest reads for initial disjointig extension stage (usually the memory bottleneck). The parameter --asm-coverage specifies the target coverage of the longest reads. Typically, 40x longest reads is enough to produce good disjointigs. Regardless of this parameter, all reads will be used at the later pipeline stages (e.g. for repeat resolution).
To polish an existing assembly, you can run Flye polisher as a standalone tool using --polish-target. Paths to reads are specified similarly to the assembly mode, and bam file could also be proveded instead of reads (the mapping stage in this case will be skipped).
Polishing is performed as the final assembly stage. By default, Flye runs one polishing iteration. Additional iterations might correct a small number of extra errors (due to improvements on how reads may align to the corrected assembly). If the parameter is set to 0, the polishing is not performed.
Use --resume to resume a previous run of the assembler that may have terminated prematurely (using the same output directory). The assembly will continue from the last previously completed step.
You might also resume from a particular stage with --resume-from stage_name, where stage_name is a choice of assembly, consensus, repeat, trestle, polishing. For example, you might supply different sets of reads for different stages.
The main output files are:
assembly.fasta - Final assembly. Contains contigs and possibly scaffolds (see below).assembly_graph.{gfa|gv} - Final repeat graph. Note that the edge sequences might be different (shorter) than contig sequences, because contigs might include multiple graph edges (see below).assembly_info.txt - Extra information about contigs (such as length or coverage).Each contig is formed by a single unique graph edge. If possible, unique contigs are extended with the sequence from flanking unresolved repeats on the graph. Thus, a contig fully contains the corresponding graph edge (with the same id), but might be longer then this edge. This is somewhat similar to unitig-contig relation in OLC assemblers. In a rare case when a repetitive graph edge is not covered by the set of “extended” contigs, it will be also output in the assembly file.
Sometimes it is possible to further order contigs into scaffolds based on the repeat graph structure. These ordered contigs will be output as a part of scaffold in the assembly file (with a scaffold_ prefix). Since it is hard to give a reliable estimate of the gap size, those gaps are represented with the default 100 Ns. assembly_info.txt file (below) contains additional information about how scaffolds were formed.
Extra information about contigs/scaffolds is output into the assembly_info.txt file. It is a tab-delimited table with the columns as follows:
Scaffold gaps are marked with ?? symbols, and * symbol denotes a terminal graph node.
Alternative contigs (representing alternative haplotypes) will have the same alt. group ID. Primary contigs are marked by *
The Flye algorithms are using repeat graph as a core data structure. In difference to de Bruijn graphs which require exact k-mer matches, repeat graphs are built using approximate sequence matches, thus can tollerate higher noise of SMS reads.
The edges of repeat graph represent genomic sequence, and nodes define the junctions. All edges are classified into unique and repetitive. The genome traverses the graph in an unknown way, so as each unique edge appears exactly once in this traversal. Repeat graphs are useful for repeat analysis and resolution - which are one of the key genome assembly challenges.
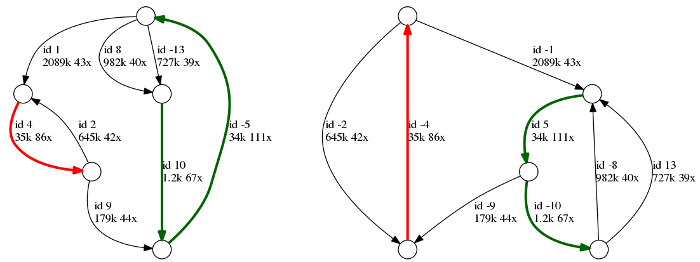
Above is an example of a repeat graph of a bacterial assembly. Each edge is labeled with its id, length and coverage. Repetitive edges are shown in color, and unique edges are black. Note that each edge is represented in two copies: forward and reverse complement (marked with +/- signs), therefore the entire genome is represented in two copies as well.
In this example, there are two unresolved repeats: (i) a red repeat of multiplicity two and length 35k and (ii) a green repeat cluster of multiplicity three and length 34k - 36k. As the repeats remained unresolved, there are no reads in the dataset that cover those repeats in full. Five unique edges will correspond to five contigs in the final assembly.
Repeat graphs produced by Flye could be visualized using AGB or Bandage.
Repeat graph before repeat resolution could be found in the 20-repeat/graph_before_rr.gv file.
| Genome | Data | Asm.Size | NG50 | CPU time | RAM |
|---|---|---|---|---|---|
| E.coli | PB 50x | 4.6 Mb | 4.6 Mb | 2 h | 2 Gb |
| C.elegans | PB 40x | 107 Mb | 2.7 Mb | 100 h | 31 Gb |
| A.thaliana | PB 75x | 120 Mb | 8.7 Mb | 100 h | 59 Gb |
| D.melanogaster | ONT 30x | 136 Mb | 13.8 Mb | 130 h | 33 Gb |
| D.melanogaster | PB 120x | 141 Mb | 11.5 Mb | 150 h | 70 Gb |
| Human NA12878 | ONT 35x (rel6) | 2.8 Gb | 30.3 Mb | 3100 h | 394 Gb |
| Human CHM13 ONT | ONT 120x (rel5) | 2.9 Gb | 69.5 Mb | 4000 h | 450 Gb |
| Human CHM13 HiFi | PB HiFi 30x | 3.0 Gb | 34.8 Mb | 780 h | 141 Gb |
| Human HG002 | PB ONT 110x | 2.9 Gb | 46.9 Mb | 4000 h | 409 Gb |
| Human CHM1 | PB 100x | 2.8 Gb | 18.6 Mb | 2700 h | 444 Gb |
| Cliveome Q20 | ONT 35x | 3.0 Gb | 46.5 Mb | 2000 h | 257 Gb |
| HMP mock | PB meta 7 Gb | 68 Mb | N/A | 60 h | 72 Gb |
| Zymo Even | ONT meta 14 Gb | 65 Mb | N/A | 60 h | 129 Gb |
| Zymo Log | ONT meta 16 Gb | 29 Mb | N/A | 100 h | 76 Gb |
| Sheep gut | HiFi meta 255G | 4.2 Gb | N/A | 3500 h | 662 Gb |
The assemblies generated using Flye 2.9 could be downloaded from Zenodo. All datasets were run with default parameters for the corresponding read type with the following exceptions: CHM13 T2T, CHM1 and HG002 were run with --asm-coverage 50.
Note that this version of the table reflects contigs NG50, while the previous versions were refering to scaffold NG50.
This is a brief description of the Flye algorithm. Please refer to the manuscript for more detailed information. The draft contig extension is organized as follows:
Note that we do not attempt to resolve repeats at this stage, thus the reconstructed contigs might contain misassemblies. Flye then aligns the reads on these draft contigs using minimap2 and calls a consensus. Afterwards, Flye performs repeat analysis as follows:
If enabled, after resolving bridged repeats, Trestle module attempts to resolve simple unbridged repeats (of multiplicity 2) using the heterogeneities between repeat copies. Finally, Flye performs polishing of the resulting assembly to correct the remaining errors:
The polishing steps could be repeated, which might slightly increase quality for some datasets.