General FAQ¶
Tip
For support or questions please post to Biostars. For bug reports and feature requests please open an issue <on github.
Note
We also have a Galaxy-related FAQ with questions that are more specific to Galaxy rather than deepTools usage.
How does deepTools handle data from paired-end sequencing?¶
Generally, all the modules working on BAM files (multiBamSummary, bamCoverage, bamCompare, plotFingerprint, computeGCBias) automatically recognize paired-end sequencing data and will use the fragment size based on the distance between read pairs.
You can by-pass the typical fragment handling on mate pairs with the option --doNotExtendPairedEnds (can be found under “advanced options” in Galaxy).
How can I test a tool with little computation time?¶
When you’re playing around with the tools to see what kinds of results they will produce, you can limit the operation to one chromosome or a specific region to save time. In Galaxy, you will find this under “advanced output options” –> “Region of the genome to limit the operation to”. The command line option is called --region (CHR:START:END).
The following tools currently have this option:
It works as follows: first, the entire genome represented in the BAM file will be regarded and sampled, then all the regions or sampled bins that do not overlap the region indicated by the user will be discarded.
Note
You can limit the operation to only one chromosome (or one specific locus on a chromosome) at a time. If you would like to limit the operation to more than one region, see the answer to the next question.
Can I specify more than one chromosome in the --regions option?¶
The short answer is: no.
Several programs allow specifying a specific regions.
For these, the input must be in the format of chr:start:end, for example “chr10” or “chr10:456700:891000”.
For these programs, it is not possible to indicate more than one region, e.g. chr10, chr11 - this will not work! Here are some ideas for workarounds if you none-the-less need to do this:
General workaround¶
Since all the tools that have the --region option work on BAM files, you could filter your reads prior to running the program, e.g. using intersectBed with --abam or samtools view. Then use the resulting (smaller) BAM file with the deepTools program of your choice.
$ samtools view -b -L regionsOfInterest.bed Reads.bam > ReadsOverlappingWithRegionsOfInterest.bam
or
$ intersectBed -abam Reads.bam -b regionsOfInterest.bed > ReadsOverlappingWithRegionsOfInterest.bam
Build-in solutions¶
computeGCBias and multiBamSummary offer build-in solutions so that you do not need to resort to tools outside of deepTools.
- multiBamSummary has two modes,
binsandBED. If you make use of the
BEDmode, you can supply a BED file of regions that you would like to limit the operation to. This will do the same thing as in the general workaround mentioned above.- computeGCBias has a
--filterOutoption. If you to create a BED file that contains all the regions you are not interested in, you can then supply this file to
computeGCBias --filterOut Regions_to_be_ignored.bedand those regions will subsequently be ignored.
When should I exclude regions from computeGCBias?¶
Note
In general, we recommend to only correct for GC bias (using computeGCBias followed by correctGCBias) if the majority of the genome (e.g., for mouse and human genomes the region between 30-60%) is GC-biased and you want to compare this sample with another sample that is not GC-biased.
Sometimes, a certain GC bias is expected, for example for ChIP samples of H3K4Me3 in mammalian samples where GC-rich promoters are expected to be enriched. To not confound the GC bias caused by the library preparation with the inherent, expected GC-bias, we incorporated the possibility to supply a file of regions to computeGCBias that will be excluded from the GC bias calculation. This file should typically contain those regions that one expects to be significantly enriched. This allows the tool to focus on background regions.
When should I use bamCoverage or bamCompare?¶
Both tools produce bigWig files, i.e. they translate the read-centered information from a BAM file into scores for genomic regions of a fixed size. The only difference is the number of BAM files that the tools use as input: while bamCoverage will only take one BAM file and produce a coverage file that is mostly normalized for sequencing depth, bamCompare will take two BAM files that can be compared with each other using several mathematical operations.
bamCompare will always normalize for sequencing depth like bamCoverage, but then it will perform additional calculations depending on what the user chose, for example:
What should I pay attention to when dealing with RNA-seq data?¶
By default, deepTools (since version 2) makes use of the information stored in the so-called CIGAR string of the alignment file (SAM/BAM specification). The CIGAR tells precisely to which bases of the reference a read maps - and, accordingly, which bases are skipped in the case of reads that span introns. These so-called split reads are natively handled by all modules of deepTools 2.0.
Warning
It is generally not recommended to activate the deepTools parameter --extendReads for RNA-seq data.
This is because there is no verified information on the fragment alignment outside the actual read sequence. A simple extension of a read over uncovered parts would probably be wrong for a lot of fragments! Activating the read extension also deactivates the utilization of the CIGAR.
How does computeMatrix handle overlapping genome regions?¶
If the BED file supplied to computeMatrix contains regions that overlap but they will just be taken as is. If you would like to prevent this, then clean the BED file before using computeMatrix. There are several methods for modifying your BED file.
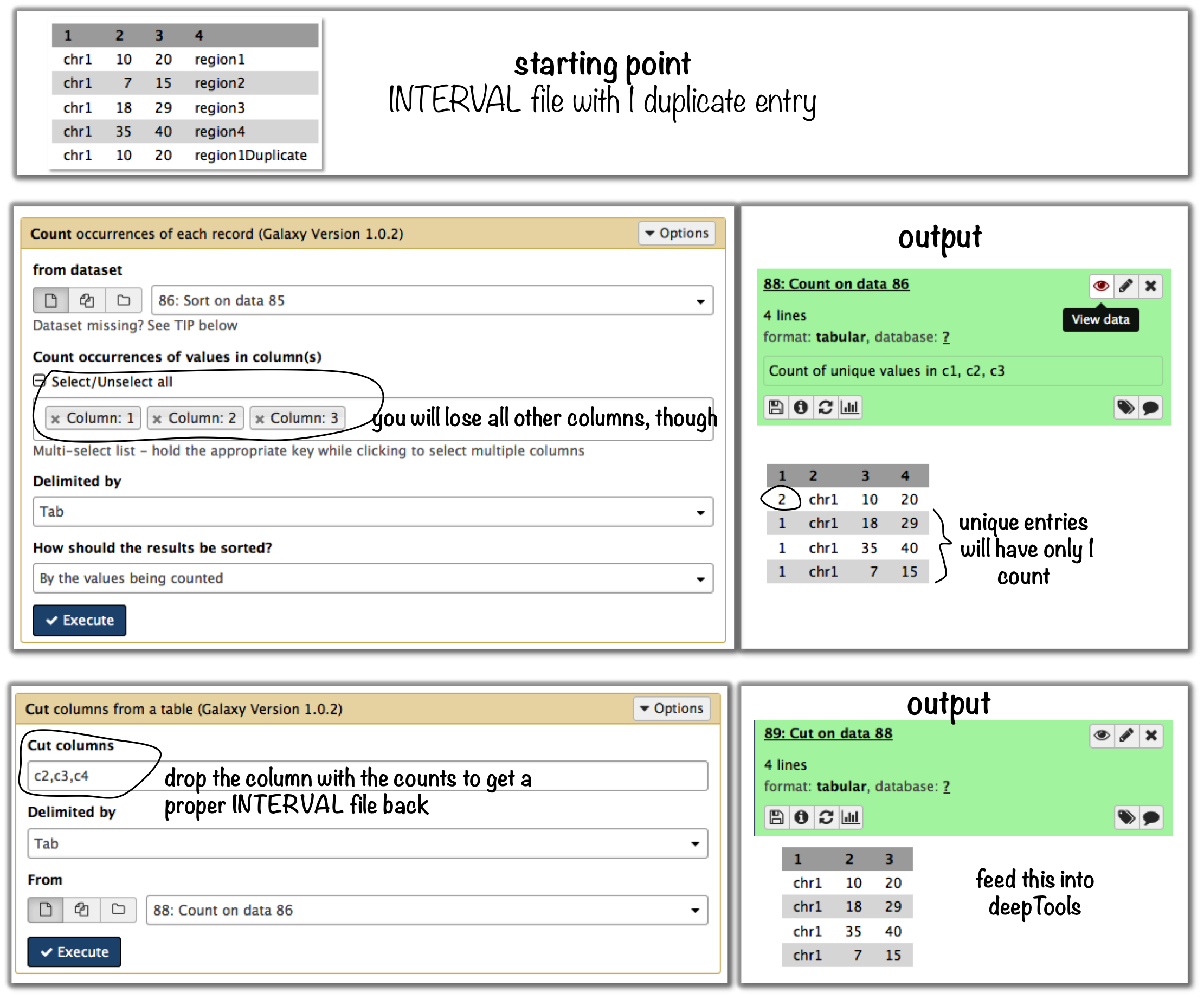
Let’s say your file looks like this:
$ cat testBed.bed
chr1 10 20 region1
chr1 7 15 region2
chr1 18 29 region3
chr1 35 40 region4
chr1 10 20 region1Duplicate
Galaxy-based work around¶
To eliminate entries with identical genome coordinates, first use the tool “Count” and then filter out all entries that are present more than once.
Command line-based work arounds¶
if you just want to eliminate identical entries (here: region1 and region1Duplicate), use
sortanduniqin the shell (note that the label of the identical regions is different - asuniqcan only ignore fields at the beginning of a file, userevto revert the sorted file, thenuniqwith ignoring the first field (which is now the name column) and then revert back:$ sort -k1,1 -k2,2n testBed.bed | rev | uniq -f1 | rev chr1 10 20 region1 chr1 7 15 region2 chr1 18 29 region3 chr1 35 40 region4
if you would like to merge all overlapping regions into one big one, use the
mergeBedfrom the BEDtools suite:again, the BED file must be sorted first
-nand-nmstellmergeBedto output the number of overlapping regions and the names of themin the resulting file, regions 1, 2 and 3 are merged
$ sort -k1,1 -k2,2n testBed.bed | mergeBed -i stdin -n -nms chr1 7 29 region2;region1;region1Duplicate;region3 4 chr1 35 40 region4 1
if you would like to keep only regions that do not overlap with any other region in the same BED file, use the same
mergeBedroutine but subsequently filter out those regions where several regions were merged.the
awkcommand will check the last field of each line ($NF) and will print the original line ($0) only if the last field contained a number smaller than 2$ sort -k1,1 -k2,2n testBed.bed | mergeBed -i stdin -n -nms | awk '$NF < 2 {print $0}' chr1 35 40 region4 1
Why does the maximum value in the heatmap not equal the maximum value in the matrix?¶
Additional processing, such as outlier removal, is done on the matrix prior to plotting the heatmap. We’ve found this beneficial in most cases. You can override this by manually setting --zMax and/or `--zMin, respectively.
The heatmap I generated looks very “coarse”, I would like a much more fine-grained image.¶
decrease the bin size when generating the matrix using computeMatrix
- In Galaxy:
go to “advanced options” –> “Length, in base pairs, of the non-overlapping bin for averaging the score over the regions length” –> define a smaller value, e.g. 50 or 25 bp
make sure that you used a sufficiently small bin size when calculating the bigWig file, though (if generated with deepTools, you can check the option “bin size”)
How can I change the automatic labels of the clusters in a k-means clustered heatmap?¶
Each cluster is treated exactly the same way as different groups of regions. Therefore, you can use the same option to define the labels of the final heatmap:
- In Galaxy:
plotHeatmap –> “Advanced output options” –> “Labels for the regions plotted in the heatmap”.
If you indicated 2 clusters for k-means clustering, enter here: C1, C2, –> instead of the full default label (“cluster 1”), the heatmap will be labeled with the abbreviations.
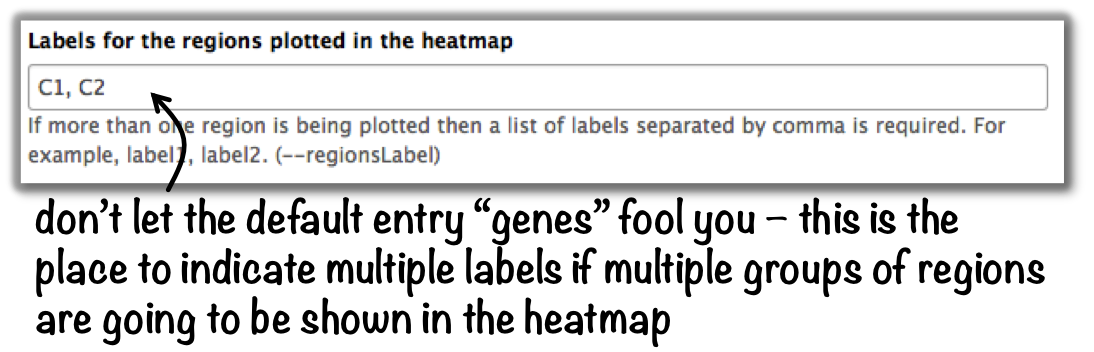
In the command line, use the --regionsLabel option to define the customized names for the regions.
How can I manually specify several groups of regions (instead of clustering)?¶
Simply specify multiple BED files (e.g., genes.bed, exons.bed and introns.bed). This works both in Galaxy and on the command line.
What do I have to pay attention to when working with a draft version of a genome?¶
If your genome isn’t included in our standard dataset then you’ll need the following:
Effective genome size - this is mostly needed for bamCoverage and bamCompare, see below for details
Reference genome sequence in 2bit format - this is needed for computeGCBias, see 2bit for details
How do I calculate the effective genome size for an organism that’s not in your list?¶
At the moment we do not provide a tool for this purpose, so you’ll have to find a solution outside of deepTools for the time being.
The “real” effective genome size is the part of the genome that is uniquely mappable. This means that the value will depend on the genome properties (how many repetitive elements, quality of the assembly etc.) and the length of the sequenced reads as 100 million 36-bp-reads might cover less than 100 million 100-bp-reads.
We currently have these options for you:
Use an GEM
Use faCount (only if you let reads be aligned non-uniquely, too!)
Use bamCoverage
Use GEM¶
There is a tool that promises to calculate the mappability for any genome given the read length (k-mer length): GEM-Mappability Calculator . According to this reply here, you can calculate the effective genome size after running this program by counting the numbers of “!” which stands for uniquely mappable regions.
Use faCount¶
If you are using bowtie2, which reports multimappers (i.e., non-uniquely mapped reads) as a default setting, you can use faCount from UCSC tools to report the total number of bases as well as the number of bases that are missing from the genome assembly indicated by ‘N’. The effective genome size would then be the total number of base pairs minus the total number of ‘N’.
Here’s an example output of faCount on D. melanogaster genome version dm3:
$ UCSCtools/faCount dm3.fa
#seq len A C G T N cpg
chr2L 23011544 6699731 4811687 4815192 6684734 200 926264
chr2LHet 368872 90881 58504 57899 90588 71000 10958
chr2R 21146708 6007371 4576037 4574750 5988450 100 917644
chr2RHet 3288761 828553 537840 529242 826306 566820 99227
chr3L 24543557 7113242 5153576 5141498 7135141 100 995078
chr3LHet 2555491 725986 473888 479000 737434 139183 89647
chr3R 27905053 7979156 5995211 5980227 7950459 0 1186894
chr3RHet 2517507 678829 447155 446597 691725 253201 84175
chr4 1351857 430227 238155 242039 441336 100 43274
chrU 10049037 2511952 1672330 1672987 2510979 1680789 335241
chrUextra 29004656 7732998 5109465 5084891 7614402 3462900 986216
chrX 22422827 6409325 4742952 4748415 6432035 90100 959534
chrXHet 204112 61961 40017 41813 60321 0 754
chrYHet 347038 74566 45769 47582 74889 104232 8441
chrM 19517 8152 2003 1479 7883 0 132
total 168736537 47352930 33904589 33863611 47246682 6368725 6650479
In this example: Total no. bp = 168,736,537 Total no. ‘N’ = 6,368,725
Warning
This method only works if multimappers are randomly assigned to their possible locations (in such cases the effective genome size is simply the number of non-N bases).
Use bamCoverage¶
If you have a sample where you expect the genome to be covered completely, e.g. from genome sequencing, a very trivial solution is to use bamCoverage with a bin size of 1 bp and the --outFileFormat option set to ‘bedgraph’. You can then count the number of non-Zero bins (bases) which will indicate the mappable genome size for this specific sample.
Use genomeCoverageBed¶
genomeCoverageBed from the BEDtools suite can be used to calculate the number of bases in the genome for which 0 overlapping reads can be found.
As described on the BEDtools website (go to genomeCov description), you need:
a file with the chromosome sizes of your sample’s organism
a position-sorted BAM file
$ bedtools genomecov -ibam sortedBAMfile.bam -g genome.size
Where can I download the 2bit genome files required for computeGCBias?¶
The 2bit files of most genomes can be found here. Search for the .2bit ending. Otherwise, fasta files can be converted to 2bit using the UCSC program faToTwoBit (available for different platforms from UCSC here).