compute sna/atom command
compute snad/atom command
compute snav/atom command
Syntax
compute ID group-ID sna/atom rcutfac rfac0 twojmax R_1 R_2 ... w_1 w_2 ... keyword values ...
compute ID group-ID snad/atom rcutfac rfac0 twojmax R_1 R_2 ... w_1 w_2 ... keyword values ...
compute ID group-ID snav/atom rcutfac rfac0 twojmax R_1 R_2 ... w_1 w_2 ... keyword values ...
ID, group-ID are documented in compute command
sna/atom = style name of this compute command
rcutfac = scale factor applied to all cutoff radii (positive real)
rfac0 = parameter in distance to angle conversion (0 < rcutfac < 1)
twojmax = band limit for bispectrum components (non-negative integer)
R_1, R_2,... = list of cutoff radii, one for each type (distance units)
w_1, w_2,... = list of neighbor weights, one for each type
zero or more keyword/value pairs may be appended
keyword = diagonal or rmin0 or switchflag
diagonal value = 0 or 1 or 2 or 3 0 = all j1, j2, j <= twojmax, j2 <= j1 1 = subset satisfying j1 == j2 2 = subset satisfying j1 == j2 == j3 3 = subset satisfying j2 <= j1 <= j rmin0 value = parameter in distance to angle conversion (distance units) switchflag value = 0 or 1 0 = do not use switching function 1 = use switching function
Examples
compute b all sna/atom 1.4 0.99363 6 2.0 2.4 0.75 1.0 diagonal 3 rmin0 0.0
compute db all sna/atom 1.4 0.95 6 2.0 1.0
compute vb all sna/atom 1.4 0.95 6 2.0 1.0
Description
Define a computation that calculates a set of bispectrum components for each atom in a group.
Bispectrum components of an atom are order parameters characterizing the radial and angular distribution of neighbor atoms. The detailed mathematical definition is given in the paper by Thompson et al. (Thompson)
The position of a neighbor atom i’ relative to a central atom i is a point within the 3D ball of radius R_ii’ = rcutfac*(R_i + R_i’)
Bartok et al. (Bartok), proposed mapping this 3D ball onto the 3-sphere, the surface of the unit ball in a four-dimensional space. The radial distance r within R_ii’ is mapped on to a third polar angle theta0 defined by,
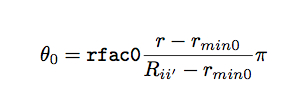
In this way, all possible neighbor positions are mapped on to a subset of the 3-sphere. Points south of the latitude theta0max=rfac0*Pi are excluded.
The natural basis for functions on the 3-sphere is formed by the 4D hyperspherical harmonics U^j_m,m’(theta, phi, theta0). These functions are better known as D^j_m,m’, the elements of the Wigner D-matrices (Meremianin, Varshalovich).
The density of neighbors on the 3-sphere can be written as a sum of Dirac-delta functions, one for each neighbor, weighted by species and radial distance. Expanding this density function as a generalized Fourier series in the basis functions, we can write each Fourier coefficient as
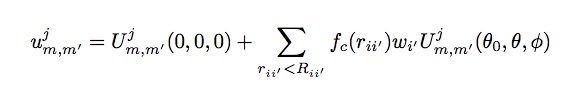
The w_i’ neighbor weights are dimensionless numbers that are chosen to distinguish atoms of different types, while the central atom is arbitrarily assigned a unit weight. The function fc(r) ensures that the contribution of each neighbor atom goes smoothly to zero at R_ii’:
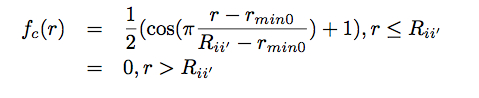
The expansion coefficients u^j_m,m’ are complex-valued and they are not directly useful as descriptors, because they are not invariant under rotation of the polar coordinate frame. However, the following scalar triple products of expansion coefficients can be shown to be real-valued and invariant under rotation (Bartok).
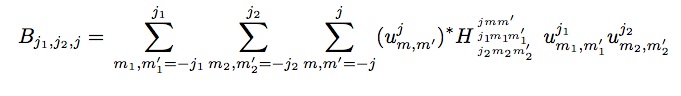
The constants H^jmm’_j1m1m1’_j2m2m2’ are coupling coefficients, analogous to Clebsch-Gordan coefficients for rotations on the 2-sphere. These invariants are the components of the bispectrum and these are the quantities calculated by the compute sna/atom. They characterize the strength of density correlations at three points on the 3-sphere. The j2=0 subset form the power spectrum, which characterizes the correlations of two points. The lowest-order components describe the coarsest features of the density function, while higher-order components reflect finer detail. Note that the central atom is included in the expansion, so three point-correlations can be either due to three neighbors, or two neighbors and the central atom.
Compute snad/atom calculates the derivative of the bispectrum components summed separately for each atom type:
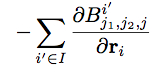
The sum is over all atoms i’ of atom type I. For each atom i, this compute evaluates the above expression for each direction, each atom type, and each bispectrum component. See section below on output for a detailed explanation.
Compute snav/atom calculates the virial contribution due to the derivatives:
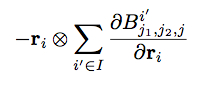
Again, the sum is over all atoms i’ of atom type I. For each atom i, this compute evaluates the above expression for each of the six virial components, each atom type, and each bispectrum component. See section below on output for a detailed explanation.
The value of all bispectrum components will be zero for atoms not in the group. Neighbor atoms not in the group do not contribute to the bispectrum of atoms in the group.
The neighbor list needed to compute this quantity is constructed each time the calculation is performed (i.e. each time a snapshot of atoms is dumped). Thus it can be inefficient to compute/dump this quantity too frequently.
The argument rcutfac is a scale factor that controls the ratio of atomic radius to radial cutoff distance.
The argument rfac0 and the optional keyword rmin0 define the linear mapping from radial distance to polar angle theta0 on the 3-sphere.
The argument twojmax and the keyword diagonal define which bispectrum components are generated. See section below on output for a detailed explanation of the number of bispectrum components and the ordered in which they are listed
The keyword switchflag can be used to turn off the switching function.
Note
If you have a bonded system, then the settings of special_bonds command can remove pairwise interactions between atoms in the same bond, angle, or dihedral. This is the default setting for the special_bonds command, and means those pairwise interactions do not appear in the neighbor list. Because this fix uses the neighbor list, it also means those pairs will not be included in the calculation. One way to get around this, is to write a dump file, and use the rerun command to compute the bispectrum components for snapshots in the dump file. The rerun script can use a special_bonds command that includes all pairs in the neighbor list.
;line
Output info:
Compute sna/atom calculates a per-atom array, each column corresponding to a particular bispectrum component. The total number of columns and the identities of the bispectrum component contained in each column depend on the values of twojmax and diagonal, as described by the following piece of python code:
for j1 in range(0,twojmax+1):
if(diagonal==2):
print j1/2.,j1/2.,j1/2.
elif(diagonal==1):
for j in range(0,min(twojmax,2*j1)+1,2):
print j1/2.,j1/2.,j/2.
elif(diagonal==0):
for j2 in range(0,j1+1):
for j in range(j1-j2,min(twojmax,j1+j2)+1,2):
print j1/2.,j2/2.,j/2.
elif(diagonal==3):
for j2 in range(0,j1+1):
for j in range(j1-j2,min(twojmax,j1+j2)+1,2):
if (j>=j1): print j1/2.,j2/2.,j/2.
Compute snad/atom evaluates a per-atom array. The columns are arranged into ntypes blocks, listed in order of atom type I. Each block contains three sub-blocks corresponding to the x, y, and z components of the atom position. Each of these sub-blocks contains one column for each bispectrum component, the same as for compute sna/atom
Compute snav/atom evaluates a per-atom array. The columns are arranged into ntypes blocks, listed in order of atom type I. Each block contains six sub-blocks corresponding to the xx, yy, zz, yz, xz, and xy components of the virial tensor in Voigt notation. Each of these sub-blocks contains one column for each bispectrum component, the same as for compute sna/atom
These values can be accessed by any command that uses per-atom values from a compute as input. See Section 6.15 for an overview of LAMMPS output options.
Restrictions
These computes are part of the SNAP package. They are only enabled if LAMMPS was built with that package. See the Making LAMMPS section for more info.
Default
The optional keyword defaults are diagonal = 0, rmin0 = 0, switchflag = 1.
(Thompson) Thompson, Swiler, Trott, Foiles, Tucker, under review, preprint available at arXiv:1409.3880
(Bartok) Bartok, Payne, Risi, Csanyi, Phys Rev Lett, 104, 136403 (2010).
(Meremianin) Meremianin, J. Phys. A, 39, 3099 (2006).
(Varshalovich) Varshalovich, Moskalev, Khersonskii, Quantum Theory of Angular Momentum, World Scientific, Singapore (1987).