Analysis steps
- Optional reference sequence correction
- Read basecall quality trimming
- Paired read merging
- Alignment to reference sequences
- Primer trimming and enforcement of read location requirements
- Ambiguously aligned mutation handling
- Multinucleotide mutation handling
- Post-alignment basecall quality filtering
- Chemical adduct location inference
- Calculation of mutation rates
- Reactivity profile calculation and normalization
- Quality control checks
Steps are listed in order of execution, with the exception of reference sequence correction, which is a larger preprocessing stage that includes many of the other listed stages.
Optional reference sequence correction
If ShapeMapper is given --correct-seq and reads from an unmodified sample, reference sequences will be automatically corrected before any SHAPE analysis. Specifically, ShapeMapper performs sequence alignment, identifies mutations occurring with frequencies above 60%, and changes the reference sequence(s) to match. Adjusted FASTA files are produced in the main output folder, and the ShapeMapper log will include messages detailing the sequence changes made and warning if other high but sub-threshold mutations are detected.
This sequence correction is useful for cases in which one major RNA species is present and contains sequence changes compared to a reference sequence (such as when studying an RNA virus whose sequence may have drifted from a reference strain). However, this is insufficient for samples containing a mixture of related but unknown sequences (such as studying an mRNA in the presence of transcribed pseudogenes of similar sequence). In such cases, see FAQ.
Details
Reads with aligner-reported mapping qualities (SAM MAPQ field) below 10 are excluded within this stage.
Within this stage, nearby mutations are not treated as larger multinucleotide mutations, and ambiguously aligned mutations are not realigned, since these operations make it more difficult to estimate the frequency of SNPs. This is different compared to how mutations are handled when estimating relative SHAPE adduct frequencies - see Multinucleotide mutation handling and Ambiguously aligned mutation handling.
Parameters
--min-seq-depth Minimum read depth required to include a mutation at a given position.
--min-freq Minimum estimated mutation frequency to include a mutation.
Example command line
shapemapper --name example --target TPP.fa --out --amplicon TPP_mutant --correct-seq --folder TPPminus --modified --folder TPPplus --untreated --folder TPPminus
Initial basecall quality trimming
Reads are scanned from left to right, and if a window of --window-to-trim nucleotides is found with less than --min-qual-to-trim average basecall quality score (phred score), that window and any nucleotides to the right are removed from the read. If the remaining read is shorter than --min-length-to-trim , it is removed (for file formatting purposes, it is actually set to a placeholder read containing one 'N', and later ignored in alignment).
The default trimming settings are intentionally somewhat permissive, and will allow reads through with some bad basecalls, especially if bad basecalls are isolated (i.e. a single bad position rather than a run of them). After alignment, a more stringent filtering step will exclude bad basecalls from contributing to identified mutations and effective read depths. See Post-alignment basecall quality filtering.
Paired read merging
If paired reads are provided as input (with --folder or with --R1 <r1.fastq> --R2 <r2.fastq> ), read pairs are merged using BBmerge v37.78 with the vstrict=t option. Unmerged reads are not thrown out.
Reads after this stage of processing can be optionally output by passing the --output-processed-reads option.
Alignment to reference sequences
Read alignment to one or more reference sequences is performed with either
Bowtie2 v2.3.4.3 (default), or
STAR v2.5.2a by passing the
--star-aligner option. Intermediate read alignment
files (*.sam) from this stage can be optionally output by passing the
--output-aligned
option.
General considerations
Choice of aligner
Both aligners produce nearly identical reactivity profiles for a SHAPE dataset from extracted E. coli ribosomal RNA, read out by MaP in randomly primed format (see SFig. 6 in Busan and Weeks, 2018).
Bowtie2 is the best choice for general use. STAR can be a good choice for long or many reference sequences (such as aligning to viral genomes, lncRNAs, or many mRNAs), and is often dramatically faster than Bowtie2 for large reference sequences. However, STAR's performance may suffer if a sizable fraction of input reads do not map to any input reference sequence. At least with the current parameters used by ShapeMapper, STAR may also be slightly less sensitive than Bowtie2 (that is, it may fail to align a small fraction of reads that "should have" aligned).
Soft clipping
Alignments are performed in local mode (also known as soft-clipped), that is, attempting to maximize alignment score over a sizable fraction of the read length, but removing positions on either end that do not agree with the reference sequence. This helps exclude bad basecalls within the first few nucleotides of sequencing and most adapter sequence. In practice, soft clipping seems to largely obviate the need for an explicit adapter trimming step (not included in ShapeMapper). Soft clipping can be disabled in ShapeMapper with the --disable-soft-clipping option, but this is not recommended.
Read length
Both STAR and Bowtie2 are short
read aligners, and will not perform well (and will very likely crash) on
extremely long reads, such as those produced by PacBio or Oxford Nanopore
sequencing platforms. This is an area for future work, and will involve
incorporating a long-read aligner into ShapeMapper. If existing long-read
alignments can be coerced into a SAM format, it may be possible to run
internals/bin/shapemapper_mutation_parser followed by internals/bin/shapemapper_mutation_counter,
but this is untested.
Paired read handling
If input reads are unpaired (--U or --unpaired-folder), all reads are aligned in unpaired mode. If input reads are paired (--R1|--R2 or --folder), merged reads are aligned in unpaired mode, and unmerged reads are aligned in paired mode.
Aligner parameters
For reference, the parameters passed to each aligner are explained here. Most of these parameters are not adjustable by the ShapeMapper user.
Bowtie2 parameters
-p with the value of ShapeMapper's --nproc (default=4).
--local --sensitive-local is used, unless the ShapeMapper parameter --disable-soft-clipping is given, in which case --end-to-end --sensitive is used.
--maxins with the value of ShapeMapper's --max-paired-fragment-length (default=800).
--ignore-quals
--no-unal
--mp 3,1 Intended to be slightly more consistent with STAR's alignment parameters.
--rdg 5,1 --rfg 5,1 Reduce penalty for gap extension, since multinucleotide deletions are often a large part of MaP signal.
--dpad 30 Allow somewhat larger deletions than bowtie2 default.
If --max-reseed and/or --max-search-depth is provided, explicit params "-D <max_search_depth> -R <max_reseed> -N 0 -L <seed_len> -i S,1,0.75" are used instead of --sensitive or --sensitive-local, where seed_len is 20 by default, or 22 if --disable-soft-clipping is provided.
Other notes:
Merged and unmerged reads are provided to bowtie2 through a single tab6-formatted input, which supports mixed paired/unpaired reads.
STAR parameters
--runThreadN with the value of ShapeMapper's --nproc parameter (default=4).
--alignEndsType Local is used, unless the ShapeMapper parameter --disable-soft-clipping is given, in which case --alignEndsType EndToEnd is used.
--scoreGap -1000000 Disable splice junction detection.
--scoreDelBase -1 --scoreInsBase -1 Reduce penalty for gap extension, since multinucleotide deletions are often a large part of MaP signal.
--outFilterMismatchNmax 999 --outFilterMismatchNoverLmax 999 Disable filtering by mismatch counts, since highly modified RNAs will produce some reads with many mutations.
--outMultimapperOrder Random --outSAMmultNmax 1 Only report one of the top alignments for a read that has an equivalent alignment score at multiple locations.
--outSAMattributes MD Include MD tag in SAM output (currently required by ShapeMapper mutation parser).
Other notes:
STAR does not appear to have an option analogous to bowtie2's --maxins, so ShapeMapper reimplements a check for concordant paired alignment in the mutation parsing stage.
To build a reference sequence index, STAR is run with --genomeSAindexNbases and --genomeChrBinNbits parameter values calculated as recommended by the STAR manual.
Some STAR runs with short reference sequences may nonetheless crash, which may require manually setting the --genomeSAindexNbases parameter to a lower value (this parameter is available through the main ShapeMapper executable). ShapeMapper can be configured to automatically rerun analyses in which STAR segfaulted by providing --rerun-on-segfault and --rerun-genomeSAindexNbase (default=3).
Since STAR does not support mixed paired/unpaired input, two separate STAR instances are run, which may increase memory requirements if especially large reference sequences are used. ShapeMapper disables STAR's shared-memory index mode by default, but it can be re-enabled by providing --star-shared-index (untested).
Primer trimming and enforcement of read location requirements
If --amplicon and/or --primers with a primers file are provided, directed primer trimming is performed. Otherwise, random primer trimming is performed, using the value of --random-primer-len.
Random primer trimming
Mutations overlapping (--random-primer-len + 1) nucleotides on the 3′ end of reads are discarded, and (--random-primer-len + 1) nucleotides are also excluded from contributing to effective read depth.
This trimming is not needed for Nextera-style preps, as the ends of cDNAs are largely digested away before sequencing.
Directed primer trimming
For amplicon datasets, reads are required to align with their ends within +/- --max-primer-offset nucleotides of expected PCR primer pairs. This can help to reduce the effects of off-target reverse transcription and PCR products (especially shorter products).
Specifically, for unpaired, paired, or merged reads, each read end is required to align within --max-primer-offset of a primer end location, and the nearest primer sites for each end are required to come from the same primer pair. For paired reads missing a mate pair, only one end is required to map near a primer (out of forward primers for reads on the forward strand and out of reverse primers for reads on the reverse strand).
Reads whose aligned locations do not meet requirements are excluded from further analysis, and the locations of these reads are shown in mapped depth plots as dashed black lines, labeled "off-target".
For reads meeting requirements, mutations overlapping either primer site are discarded, and primer sites are excluded from contributing to effective read depth.
Usage
For most users, simply set primer binding sites to lowercase sequence in the input .fa file, and run ShapeMapper with the --amplicon option. This will handle cases with a single pair of primers on either end of the target.
For more complex cases with internal PCR primer locations or multiple pairs of primers, run ShapeMapper with a primers file input with --primers <primers_file> . ShapeMapper will automatically determine the expected locations of input PCR primers by comparing their sequences with those of reference targets.
Excluding reads aligning to multiple locations
Reads with aligner-reported MAPQ values below --min-mapq are excluded from further analysis. The mapped locations of these excluded reads are shown in mapped depth plots as dotted red lines, annotated "low MAPQ".
If multiple RNA targets share much of their sequence, low MAPQ reads typically lack enough sequence unique to one particular target. In other cases, low MAPQ reads are simply short, low complexity reads that spuriously align to multiple homopolymeric portions of a target sequence.
Note: When using Bowtie2, high numbers of mutations in a read will result in lower aligner-reported mapping qualities. Therefore, raising the value of --min-mapq may have the side effect of excluding highly mutated reads.
Mapped depth plots
ShapeMapper produces *_mapped_depths.pdf files in the main output folder
to visualize the effects of this analysis stage and, with directed primers,
to break down the contributions of each primer pair to overall read depth.
Example plots:
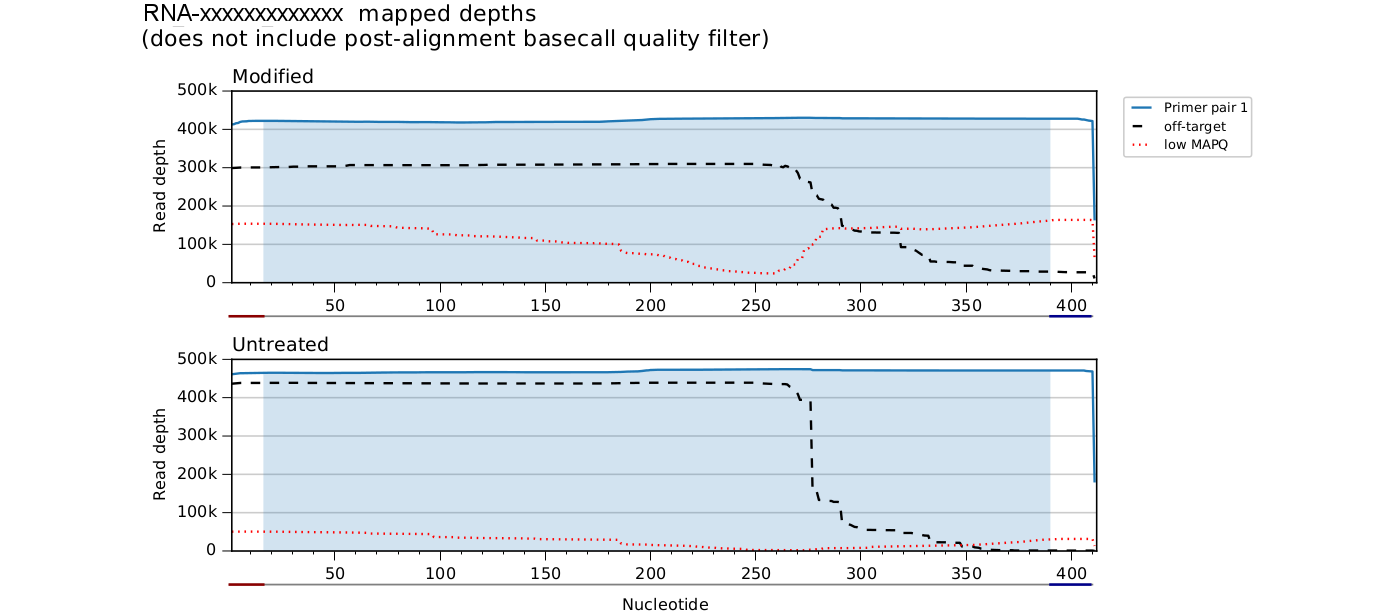
Only reads plotted with solid lines are used in further analysis, and only the shaded portion of each amplicon is included.
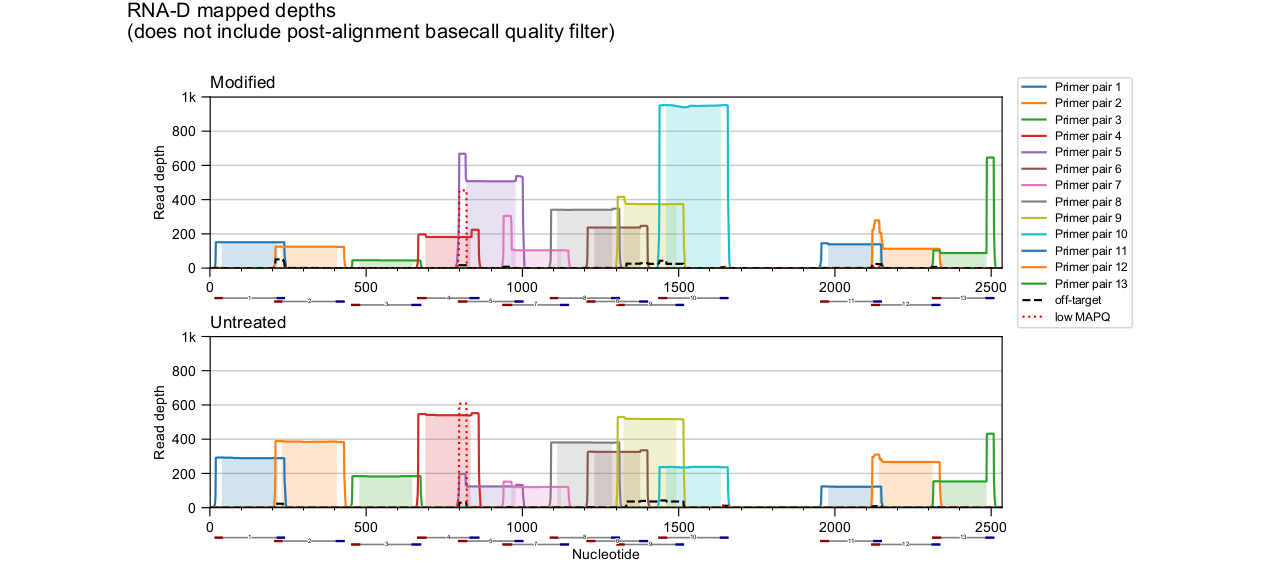
Multiple, even overlapping primer pairs are supported. Note in the above plot that apparent primer dimer or leftover primer reads are excluded - see for example the unshaded regions of the green rightmost primer pair 13.
Post-alignment paired read merging
For paired reads that fail to merge before alignment, but where both mates align to the same target for a fragment size <= --max-paired-fragment-length , ShapeMapper merges the mates into a single read, resolving any sequence or mutation conflicts attempting to favor higher-quality basecalls.
Basecall quality score resolution
At a given position where mate pairs overlap, the higher basecall quality score is used for the merged read.
Mutation resolution
Mutations that overlap are first grouped. For each group of mutations, the mean phred score over the set of basecalls within and adjacent to R1 mutations is compared to the mean phred score over R2 mutations and adjacent positions. The read mutation group with the higher mean phred score is then used for the merged read. This strategy avoids rare cases in which a more naïve mutation resolution algorithm results in a merged read that is incompatible with both mate pairs.
Ambiguously aligned mutation handling
Early versions of ShapeMapper entirely excluded ambiguously aligned mutations from analysis, since they make it impossible to infer adduct location with full single nucleotide accuracy. However, short ambiguous deletions are a fairly large signal within many MaP datasets, and we find that realigning ambiguous mutations 5′ and including them in analysis produces empirical improvements in reactivity profile accuracy. See SFig. 2 in Busan and Weeks, 2018. The default behavior (5′ realignment) can be reversed (3′ realignment) by passing the --right-align-ambig option (not recommended).
Example realignment: A five-nucleotide deletion can be equivalently aligned to two locations. ShapeMapper uses the 5′ location.
3′ aligned read: GAGGAAGGTGGGGATGACGTCA-----TCATGGCCCTTACG
5′ aligned read: GAGGAAGGTGGGGATGAC-----GTCATCATGGCCCTTACG
target: GAGGAAGGTGGGGATGACGTCAAGTCATCATGGCCCTTACG
^
inferred adduct site
We hypothesize that most short ambiguous deletions are a result of reverse transcriptase encountering an adduct, then stalling until a few nucleotides of the cDNA strand locally melt and reanneal to another complementary position within the RNA a few nucleotides toward the 5′ end.
Repeat and/or homopolymeric regions still pose difficulties for MaP, both due to high background reverse transcriptase mutagenesis within these regions and due to frequent loss of information regarding adduct position. Future ShapeMapper versions may track and warn the user about such regions.
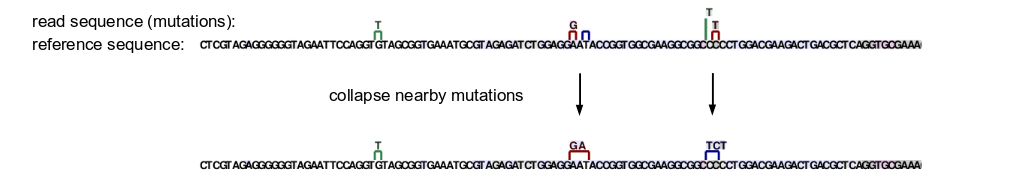
Multinucleotide mutation handling
We observe that SuperScript II reverse transcription under MaP conditions on SHAPE-modified RNA produces some reads with multiple nearby mutations, and hypothesize that many of these mutation clusters in fact reflect single adducts toward the 3′ end of a given cluster. ShapeMapper therefore applies a mutation separation threshold to collapse nearby mutations.
Mutations separated by less than --min-mutation-separation unchanged reference sequence nucleotides are collapsed and treated as a single mutation. This is not applied within sequence correction stages if present ( --correct-seq samples). The default mutation separation threshold (6 nucleotides) was empirically selected to maximize reactivity profile accuracy for an extracted E. coli rRNA SHAPE dataset (see SFig. 1 in Busan and Weeks, 2018). The same threshold also appears optimal for DMS adducts (unpublished data). Alternative reverse transcriptases, reverse transcription conditions, or adduct chemistries may require re-optimization.
Example:
Post-alignment basecall quality filtering
Initial read quality trimming is fairly permissive to support high read depths and to permit reads with a few bad basecalls to be used as long as bad basecalls are isolated (see Initial basecall quality trimming).
After alignment, ShapeMapper applies a separate, more stringent basecall quality filter to remove mutations and read depths associated with basecalls with quality scores below --min-qual-to-count. For sequencing runs of mixed quality (such as runs approaching the length limits of Illumina chemistry), this strategy enables recovery of high read depths without sacrificing reactivity profile accuracy. See SFig. 1 in Busan and Weeks, 2018.
Specifically, for each nucleotide position not within mutations (that is, a position covered by a read but unchanged from reference sequence), a given position is excluded if its basecall, the basecall immediately 5′, or the basecall immediately 3′ has a quality score below --min-qual.
A given mutation is excluded (and the covered sequence region) if it contains any basecall with quality score below --min-qual, or if the basecall immediately 5′ or the basecall immediately 3′ has a quality score below --min-qual.
Example 1:

A poorly scoring portion of the read is excluded from analysis.
Example 2:
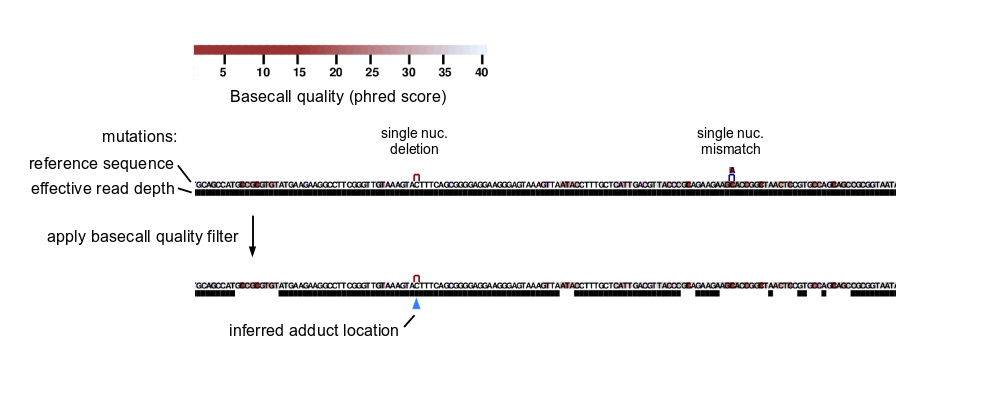
Poorly scoring regions of the read are excluded, and one mutation associated with low-quality basecalls is excluded.
Chemical adduct location inference
ShapeMapper infers the locations of chemical adducts as the reference sequence position immediately 5′ of the last unchanged reference nucleotide before a mutation scanning 3′→5′ (as in reverse transcription).
Examples:
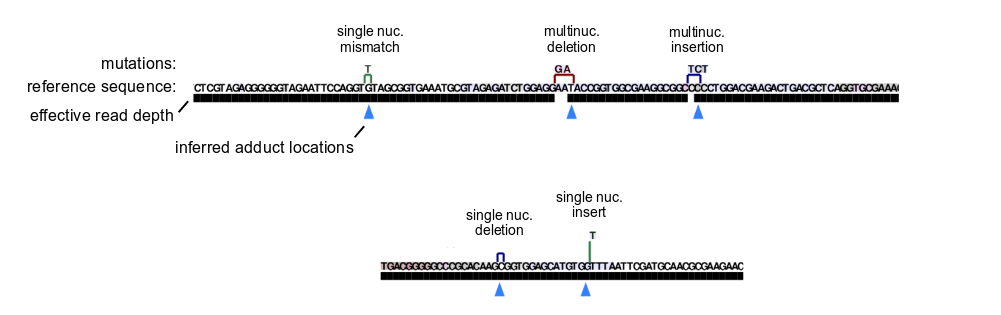
Calculation of mutation rates
Mutation and read depth counting
After all mutation and read depth processing and filtering steps are performed, ShapeMapper sums mutation counts and read depths for each position in each RNA sequence.
Effective read depth
Simple sequencing read depths are computed as the number of reads crossing a given position in the target sequence. For computing mutation rates, however, these depths are somewhat inadequate. The mutation rate at a given position should be computed as the number of observed mutations divided by the total number of observations (i.e. the number of "no mutation" observations plus the number of "mutation" observations).
A given position within the region covered by a multinucleotide mutation is not actually an observation of "no mutation" - we are effectively blind over these regions in individual reads. To help account for this, the region over each mutation is excluded from contributing to the effective read depth, with the exception of the inferred adduct site (see examples above). This correction is not applied to sequence correction stages. In practice, excluding multinucleotide sites from read depths is usually a small change to a large denominator, and has a negligable effect on reactivity profile accuracy, at least with mutation rates in the range produced by SHAPE adducts.
Effective read depths as reported by ShapeMapper also exclude low quality basecalls (see Post-alignment basecall quality filtering).
Optional outputs:
Intermediate parsed and processed mutations associated with individual reads can be optionally output by passing the --output-parsed-mutations option (see file format).
Tables of mutation counts broken down by mutation classification can be optionally output by passing the --output-counted-mutations (see file format).
Tabular histograms of read lengths and per-read mutation counts can be optionally output in the main ShapeMapper log by passing the --per-read-histograms.
Rate calculation
At each position in a given RNA, mutation rate
for each provided sample is calculated as the mutation count
divided by the effective read depth at that position. In
output tab-delimited *_profile.txt files, the relevant
columns are <sample>_mutations (mutation counts),
<sample>_effective_depth (effective read depths), and
<sample>_rate (mutation rate).
Output plots:
*_profiles.pdf output files include panels for read depths and
mutation rates.
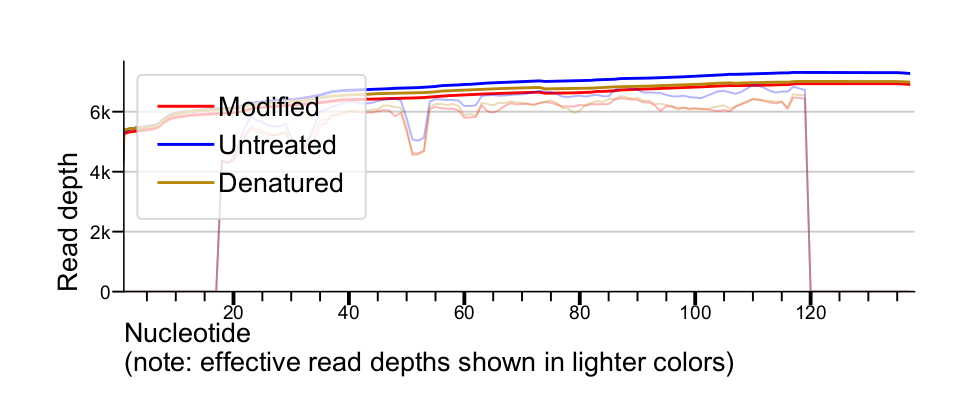
Read depths for each sample are plotted with solid lines, and effective read depths are plotted with light lines.
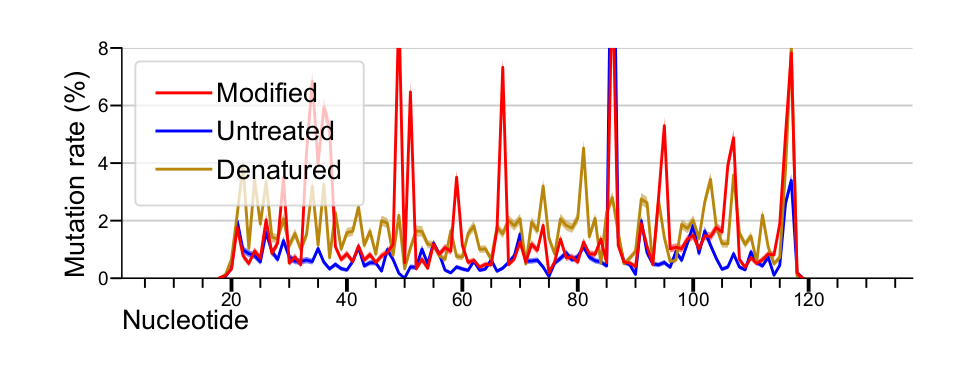
Mutation rates plotted with
lines, and standard errors plotted as shaded areas. Mutation rate standard
errors are calculated as sqrt(mutation rate) / sqrt(effective depth).
Reactivity profile calculation and normalization
Sample key: M: Modified, U: Untreated control, D: Denatured control
Reactivity
If three samples are provided, reactivity is calculated as
(rateM - rateU) / rateD
If two samples are provided, reactivity is calculated as
rateM - rateU
If only one sample is provided, reactivity is simply
rateM
Standard error
If three samples are provided, reactivity stderr is calculated as
sqrt( stderrM / rateD)2 + (stderrU / rateD)2 + (stderrD × (rateM - rateU) / rateD2)2 )
where rate and rate stderr are calculated as above.
If two samples are provided, reactivity stderr is calculated as
sqrt(stderrM2 + stderrU2)
If only one sample is provided, reactivity stderr is simply
stderrM
Reactivity standard errors are shown as error bars in *_profile.pdf plots.
Excluded positions
Lowercase sequence is excluded from reactivity profiles. Positions with effective read depth in any sample below --min-depth (default=5000) are excluded. If an untreated control sample is provided, positions with untreated mutation rate above --max-bg (default=0.05) are excluded.
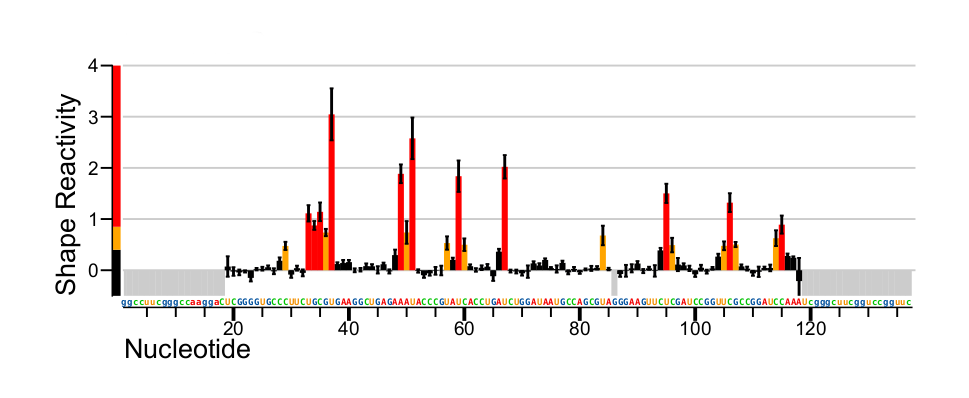
Normalization
Over the set of all RNAs and nucleotide positions without masked (lowercase) sequence, high background, or low read depth, reactivities are normalized by dividing by the mean reactivity of the top 10% of reactivities after reactivities above a threshold are excluded (see section 3.2.1 in Low and Weeks, 2010). That threshold is selected from the largest value out of
[1.5 × interquartile range,
90th percentile (if total seq length > 100) or 95th percentile (if total seq length < 100)].
By default, ShapeMapper normalizes all profiles together - that is, using the combined set of reactivities to compute a single normalization factor that is applied to all profiles. This can be disabled by passing the --indiv-norm option.
Example:
Limitations:
SHAPE-MaP measures relative probe reactivities, and normalization is an attempt to put these relative values on a more meaningful scale, closer to what might ideally be achieved with an absolute measurement of nucleotide conformational state. However, this normalization relies on the assumption that the distribution of observed reactivities in a particular experiment matches the distribution of reactivities observed in experiments used to develop pseudo-free energy structure modeling constraint parameters. In particular, normalization will produce systemically incorrect results for highly single-stranded RNAs or extremely highly base-paired RNAs.
Future work may attempt to fit relative reactivity distributions to empirical models of the distributions of reactivities observed for paired and unpaired nucleotides. This would allow more accurate structure modeling for highly structured or highly unstructured RNAs as well as providing an estimate of overall base pairing percentage.
Quality control checks
ShapeMapper performs several basic quality-control checks for each reactivity profile. These are necessarily heuristic, since different downstream analyses require different levels of data quality, and since RNAs of differing flexibility will show different overall signal levels above background. In general, more sequencing read depth is always helpful, as are higher modification/mutation rates (to a point).
If ShapeMapper gives a red warning message about possible low-quality reactivity profiles, read the log file to see which quality control checks failed, and refer to this section for possible remedies.
Read depth check
ShapeMapper requires that at least 80% of non-masked nucleotides meet a minimum sequencing depth of 5000 in all provided samples. Note that with uneven sequencing coverage, some regions of an RNA may have higher-quality reactivity data than other regions. For example, coverage is often lower near transcript ends.
If read depths are low, check alignment stats in the logfile to see the amount of target sequence present in each sample. Better target enrichment or recovery and/or additional sequencing can often help.
Positive mutation rates above background check
If an untreated control sample is provided (highly recommended), ShapeMapper requires that at least 50% of non-masked nucleotides with depths above 5000 have a higher mutation rate in the modified sample than in the untreated sample.
High background mutation rates check
If an untreated control sample is provided, ShapeMapper requires that no more than 5% of non-masked nucleotide with depths above 5000 have an untreated mutation rate above 0.05.
An unusual number of high-background nucleotides can result from the presence of native modifications or, more often, a subpopulation of sequence variants or transcribed pseudogenes. There is no built-in method to automatically determine the sequences of multiple sequence variants in a mixture, but it can sometimes be helpful to pass the --render-mutations option and visually inspect the PDF outputs from the untreated sample, looking for repeated instances of the same mutation pattern from distinct reads. By default, these outputs are limited to 100 pages, controlled with the --max-pages parameter. The pages are wide enough by default to accomodate a maximum paired read length of 800 nts, but this is often very zoomed out; lower the --max-paired-fragment-length as needed for visualization.
Number of highly reactive nucleotides check
ShapeMapper requires that at least 8% of non-masked nucleotides with depths above 5000 have a modified mutation rate above 0.006 after background subtraction.
Possible causes for failure:
- DNA contamination. Unusually low background mutation rates can be a secondary indication that this is the problem (since reverse transcription under MaP conditions usually generates some errors).
- Poor mixing of chemical reagents and RNA and/or poor reagent diffusion (if modifying in cells), resulting in low modification rates
- Expired reagents, resulting in low modification rates
- Poor reverse transcription conditions, resulting in low adduct read-through
- Extremely highly structured RNA. A molecule that genuinely contains no flexible nucleotides is indistinguishable from a highly flexible one that was mistakenly unmodified (they will both have low mutation rates above background). In this case, additional control experiments or complementary techniques may be needed.