The Program RNAduplex
Introduction
If the sequences are very long (many kb) RNAcofold is too slow
to be useful. The RNAduplex program is a fast alternative, that works by
predicting only intermolecular base pairs. It’s almost as fast as simple
sequence alignment, but much more accurate than a BLAST search.
The example below searches the 3’ UTR of an mRNA for a miRNA binding site.
Binding site prediction with RNAduplex
The file duplex.seq contains the 3’UTR of NM_024615 and the
microRNA mir-145.
$ RNAduplex < duplex.seq
>NM_024615
>hsa-miR-145
.(((((.(((...((((((((((.&)))))))))))))))))). 34,57 : 1,19 (-21.90)
Most favorable binding has an interaction energy of -21.90 kcal/mol and pairs up on positions 34-57 of the UTR with positions 1-22 of the miRNA.
RNAduplex can also produce alternative binding sites, e.g. running
RNAduplex -e 10 would list all binding sites within 10 kcal/mol of
the best one.
Since RNAduplex forms only intermolecular pairs, it neglects the
competition between intramolecular folding and hybridization. Thus, it is
recommended to use RNAduplex as a pre-filter and analyse good
RNAduplex hits additionally with RNAcofold or
RNAup. Using the example above, running RNAup will yield:
Binding site prediction with RNAup
$ RNAup -b < duplex.seq
>NM_024615
>hsa-miR-145
(((((((&))))))) 50,56 : 1,7 (-8.41 = -9.50 + 0.69 + 0.40)
GCUGGAU&GUCCAGU
RNAup output in file: hsa-miR-145_NM_024615_w25_u1.out
The free energy of the duplex is -9.50 kcal/mol and shows a discrepancy to the
structure and energy value computed by RNAduplex (differences may arise from
the fact that RNAup computes partition functions rather than optimal
structures).
However, the total free energy of binding is less favorable (-8.41 kcal/mol),
since it includes the energetic penalty for opening the binding site on the
mRNA (0.69 kcal/mol) and miRNA (0.40 kcal/mol). The -b option includes the
probability of unpaired regions in both RNAs.
You can also run RNAcofold on the example to see the complete structure
after hybridization (neither RNAduplex nor RNAup produce structure
drawings). Note however, that the input format for RNAcofold is different.
An input file suitable for RNAcofold has to be created from the
duplex.seq file first (use any text editor).
As a more difficult example, let’s look at the interaction of the bacterial
smallRNA RybB and its target mRNA ompN. First we’ll try predicting the binding
site using RNAduplex:
$ RNAduplex < RybB.seq
>RybB
>ompN
.((((..((((((.(((....((((((((..(((((.((..((.((....((((..(((((((((((..((((((&
.))))))..))))))).)))).....))))....)).)).)).))).))..))))........))))..))).)))))).)))).
5,79 : 80,164 (-34.60)
Note, that the predicted structure spans almost the full length of the RybB small RNA. Compare the predicted interaction to the structures predicted for RybB and ompN alone, and ask yourself whether the predicted interaction is indeed plausible.
Below the structure of ompN on the left and RybB on the right side. The respective binding regions predicted by RNAduplex are marked in red:
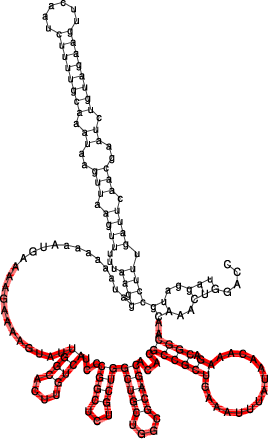

GCCAC-----TGCTTTTCTTTGATGTCCCCATTTT-GTGGA-------GC-CCATCAACCCCGCCATTTCGGTT---CAAG-GTTGGTGGGTTTTTT
||| |||| |||||| ||| ||||| |||| || ||| || || || |||| |||| || ||| |||||| -40.30
AGGTCAAACAACGGC-AGAAACAATATT--TAAAGTCGCCGCACACGACGCGGTCGTCGGT-CGTCTCGGCCCTACTGTTCACGGTTATGAAAAGAAACC-3'
Compare the RNAduplex prediction with the interaction predicted by
RNAcofold, RNAup and the handcrafted prediction you see above.