complement¶
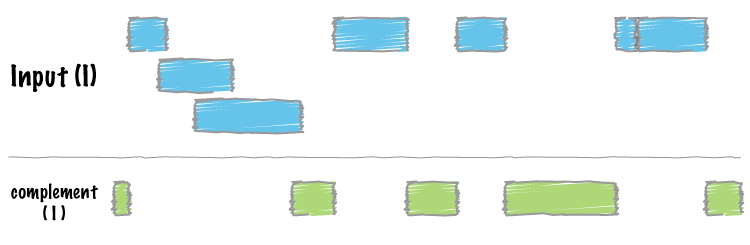
bedtools complement returns all intervals in a genome that are not
covered by at least one interval in the input BED/GFF/VCF file.
See also
Usage and option summary¶
Usage:
bedtools complement -i <BED/GFF/VCF> -g <GENOME>
(or):
complementBed -i <BED/GFF/VCF> -g <GENOME>
Default behavior¶
By default, bedtools complement returns all genomic intervals that are not
covered by at least one record from the input file.
$ cat A.bed
chr1 100 200
chr1 400 500
chr1 500 800
$ cat my.genome
chr1 1000
chr2 800
$ bedtools complement -i A.bed -g my.genome
chr1 0 100
chr1 200 400
chr1 800 1000
chr2 0 800
-L Only report chromosomes that are in the -i file¶
Use the "-L" option to L`imit the output to solely the chromosomes that are represented in the `-i file. Chromosomes that are in -g but not -i will be suppressed
For example (note the difference in coverage with and without -s:
$ cat A.bed
chr1 100 200
chr1 400 500
chr1 500 800
$ cat my.genome
chr1 1000
chr2 800
$ bedtools complement -i A.bed -g my.genome
chr1 0 100
chr1 200 400
chr1 800 1000
