slop¶
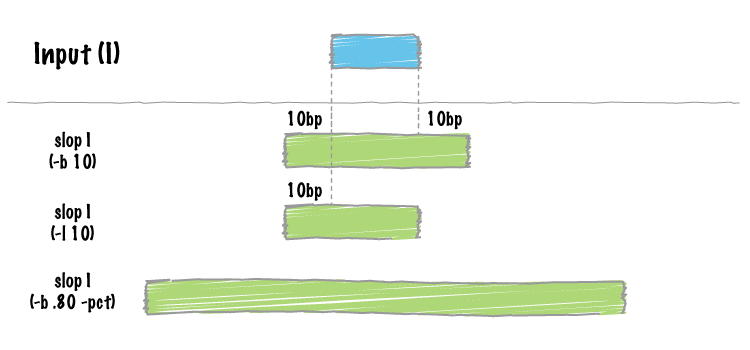
bedtools slop will increase the size of each feature in a feature file by a
user-defined number of bases. While something like this could be done with an
awk '{OFS="\t" print $1,$2-<slop>,$3+<slop>}',
bedtools slop will restrict the resizing to the size of the chromosome
(i.e. no start < 0 and no end > chromosome size).
Note
In order to prevent the extension of intervals beyond chromosome boundaries,
bedtools slop requires a genome file defining the length of each
chromosome or contig.
See also
Usage and option summary¶
Usage:
bedtools slop [OPTIONS] -i <BED/GFF/VCF> -g <GENOME> [-b or (-l and -r)]
(or):
slopBed [OPTIONS] -i <BED/GFF/VCF> -g <GENOME> [-b or (-l and -r)]
Option |
Description |
|---|---|
-b |
Increase the BED/GFF/VCF entry by the same number base pairs in each direction. Integer. |
-l |
The number of base pairs to subtract from the start coordinate. Integer. |
-r |
The number of base pairs to add to the end coordinate. Integer. |
-s |
Define -l and -r based on strand. For example. if used, -l 500 for a negative-stranded feature, it will add 500 bp to the end coordinate. |
-pct |
Define -l and -r as a fraction of the feature's length. E.g. if used on a 1000bp feature, -l 0.50, will add 500 bp "upstream". Default = false. |
-header |
Print the header from the input file prior to results. |
Default behavior¶
By default, bedtools slop will either add a fixed number of bases in each
direction (-b) or an asymmetric number of bases in each direction
with -l and -r.
$ cat A.bed
chr1 5 100
chr1 800 980
$ cat my.genome
chr1 1000
$ bedtools slop -i A.bed -g my.genome -b 5
chr1 0 105
chr1 795 985
$ bedtools slop -i A.bed -g my.genome -l 2 -r 3
chr1 3 103
chr1 798 983
However, if the requested number of bases exceeds the boundaries of the
chromosome, bedtools slop will "clip" the feature accordingly.
$ cat A.bed
chr1 5 100
chr1 800 980
$ cat my.genome
chr1 1000
$ bedtools slop -i A.bed -g my.genome -b 5000
chr1 0 1000
chr1 0 1000
-s Resizing features according to strand¶
bedtools slop will optionally increase the size of a feature based on strand.
For example:
$ cat A.bed
chr1 100 200 a1 1 +
chr1 100 200 a2 2 -
$ cat my.genome
chr1 1000
$ bedtools slop -i A.bed -g my.genome -l 50 -r 80 -s
chr1 50 280 a1 1 +
chr1 20 250 a2 2 -
-pct Resizing features by a given fraction¶
bedtools slop will optionally increase the size of a feature by a
user-specific fraction.
For example:
$ cat A.bed
chr1 100 200 a1 1 +
$ bedtools slop -i A.bed -g my.genome -b 0.5 -pct
chr1 50 250 a1 1 +
$ bedtools slop -i a.bed -l 0.5 -r 0.0 -pct -g my.genome
chr1 50 200 a1 1 +
-header Print the header for the A file before reporting results.¶
By default, if your A file has a header, it is ignored when reporting results. This option will instead tell bedtools to first print the header for the A file prior to reporting results.
