Utilitary functions for codon sites. More...
#include <Bpp/Seq/CodonSiteTools.h>
 Inheritance diagram for bpp::CodonSiteTools:
Inheritance diagram for bpp::CodonSiteTools: Collaboration diagram for bpp::CodonSiteTools:
Collaboration diagram for bpp::CodonSiteTools:
Public Member Functions | |
| CodonSiteTools () | |
| virtual | ~CodonSiteTools () |
Static Public Member Functions | |
| static bool | hasGapOrStop (const Site &site, const GeneticCode &gCode) throw (AlphabetException) |
| Method to know if a codon site contains gap(s) or stop codons. More... | |
| static bool | hasStop (const Site &site, const GeneticCode &gCode) throw (AlphabetException) |
| Method to know if a codon site contains stop codon or not. More... | |
| static bool | isMonoSitePolymorphic (const Site &site) throw (AlphabetException, EmptySiteException) |
| Method to know if a polymorphic codon site is polymorphic at only one site. More... | |
| static bool | isSynonymousPolymorphic (const Site &site, const GeneticCode &gCode) throw (AlphabetException, AlphabetMismatchException, EmptySiteException) |
| Method to know if polymorphism at a codon site is synonymous. More... | |
| static Site * | generateCodonSiteWithoutRareVariant (const Site &site, const GeneticCode &gCode, double freqmin) throw (AlphabetException, EmptySiteException) |
| generate a codon site without rare variants More... | |
| static size_t | numberOfDifferences (int i, int j, const CodonAlphabet &ca) |
| Compute the number of differences between two codons. More... | |
| static double | numberOfSynonymousDifferences (int i, int j, const GeneticCode &gCode, bool minchange=false) |
| Compute the number of synonymous differences between two codons. More... | |
| static double | piSynonymous (const Site &site, const GeneticCode &gCode, bool minchange=false) throw (AlphabetException, AlphabetMismatchException, EmptySiteException) |
| Compute the synonymous pi per codon site. More... | |
| static double | piNonSynonymous (const Site &site, const GeneticCode &gCode, bool minchange=false) throw (AlphabetException, AlphabetMismatchException, EmptySiteException) |
| Compute the non-synonymous pi per codon site. More... | |
| static double | numberOfSynonymousPositions (int i, const GeneticCode &gCode, double ratio=1.0) throw (Exception) |
| Return the number of synonymous positions of a codon. More... | |
| static double | meanNumberOfSynonymousPositions (const Site &site, const GeneticCode &gCode, double ratio=1) throw (AlphabetException, AlphabetMismatchException, EmptySiteException) |
| Return the mean number of synonymous positions per codon site. More... | |
| static size_t | numberOfSubsitutions (const Site &site, const GeneticCode &gCode, double freqmin=0.) throw (AlphabetException, EmptySiteException) |
| Return the number of subsitutions per codon site. More... | |
| static size_t | numberOfNonSynonymousSubstitutions (const Site &site, const GeneticCode &gCode, double freqmin=0.) throw (AlphabetException, AlphabetMismatchException, EmptySiteException) |
| Return the number of Non Synonymous subsitutions per codon site. More... | |
| static std::vector< size_t > | fixedDifferences (const Site &siteIn, const Site &siteOut, int i, int j, const GeneticCode &gCode) throw (AlphabetException, AlphabetMismatchException, EmptySiteException) |
| Return a vector with the number of fixed synonymous and non-synonymous differences per codon site. More... | |
| static bool | isFourFoldDegenerated (const Site &site, const GeneticCode &gCode) |
| static void | getCounts (const SymbolList &list, std::map< int, size_t > &counts) |
| Count all states in the list. More... | |
| static void | getCounts (const SymbolList &list1, const SymbolList &list2, std::map< int, std::map< int, size_t > > &counts) throw (DimensionException) |
| Count all pair of states for two lists of the same size. More... | |
| static void | getCounts (const SymbolList &list, std::map< int, double > &counts, bool resolveUnknowns) |
| Count all states in the list, optionaly resolving unknown characters. More... | |
| static void | getCounts (const SymbolList &list1, const SymbolList &list2, std::map< int, std::map< int, double > > &counts, bool resolveUnknowns) throw (DimensionException) |
| Count all pair of states for two lists of the same size, optionaly resolving unknown characters. More... | |
| static void | getFrequencies (const SymbolList &list, std::map< int, double > &frequencies, bool resolveUnknowns=false) |
| Get all states frequencies in the list. More... | |
| static void | getFrequencies (const SymbolList &list1, const SymbolList &list2, std::map< int, std::map< int, double > > &frequencies, bool resolveUnknowns=false) throw (DimensionException) |
| Get all state pairs frequencies for two lists of the same size.. More... | |
| static double | getGCContent (const SymbolList &list, bool ignoreUnresolved=true, bool ignoreGap=true) throw (AlphabetException) |
| Get the GC content of a symbol list. More... | |
| static size_t | getNumberOfDistinctPositions (const SymbolList &l1, const SymbolList &l2) throw (AlphabetMismatchException) |
| Get the number of distinct positions. More... | |
| static size_t | getNumberOfPositionsWithoutGap (const SymbolList &l1, const SymbolList &l2) throw (AlphabetMismatchException) |
| Get the number of positions without gap. More... | |
| static void | changeGapsToUnknownCharacters (SymbolList &l) |
| Change all gap elements to unknown characters. More... | |
| static void | changeUnresolvedCharactersToGaps (SymbolList &l) |
| Change all unknown characters to gap elements. More... | |
Detailed Description
Utilitary functions for codon sites.
Definition at line 60 of file CodonSiteTools.h.
Constructor & Destructor Documentation
◆ CodonSiteTools()
|
inline |
Definition at line 64 of file CodonSiteTools.h.
◆ ~CodonSiteTools()
|
inlinevirtual |
Definition at line 65 of file CodonSiteTools.h.
Member Function Documentation
◆ changeGapsToUnknownCharacters()
|
staticinherited |
Change all gap elements to unknown characters.
- Parameters
-
l The input list of characters.
Definition at line 180 of file SymbolListTools.cpp.
References bpp::SymbolList::getAlphabet(), bpp::Alphabet::getUnknownCharacterCode(), bpp::Alphabet::isGap(), and bpp::SymbolList::size().
◆ changeUnresolvedCharactersToGaps()
|
staticinherited |
Change all unknown characters to gap elements.
- Parameters
-
l The input list of characters.
Definition at line 189 of file SymbolListTools.cpp.
References bpp::SymbolList::getAlphabet(), bpp::Alphabet::getGapCharacterCode(), bpp::Alphabet::isUnresolved(), and bpp::SymbolList::size().
◆ fixedDifferences()
|
static | ||||||||||||||||||||||||||||||||||||||||
Return a vector with the number of fixed synonymous and non-synonymous differences per codon site.
Compute the number of synonymous and non-synonymous differences between the concensus codon of SiteIn (i) and SiteOut (j), which are fixed within each alignement. Example:
Here, the first position is non-synonymous different and fixed, the third position is synonymous different but not fixed (polymorphic in SiteIn). The return vector is thus [0,1]. In case of complex codon, the path that gives the minimum number of non-synonymous changes is chosen. The argument minchange=true is sent to numberOfSynonymousDifferences used in this method. Otherwise, a non-integer number could be return.
Rare variants (<= freqmin) can be excluded.
- Parameters
-
siteIn a Site siteOut a Site i an integer j an integer gCode a GeneticCode
- Exceptions
-
AlphabetException If the alphabet associated to one of the sites is not a codon alphabet. AlphabetMismatchException If the codon alphabet each the site do not match the codon alphabet of the genetic code. EmptySiteException If one of the sites has size 0.
Definition at line 682 of file CodonSiteTools.cpp.
References bpp::CodonAlphabet::getFirstPosition(), bpp::CodonAlphabet::getNucleicAlphabet(), bpp::CodonAlphabet::getSecondPosition(), bpp::CodonAlphabet::getThirdPosition(), bpp::AlphabetTools::isCodonAlphabet(), and bpp::SiteTools::isConstant().
◆ generateCodonSiteWithoutRareVariant()
|
static | ||||||||||||||||||||||||||||
generate a codon site without rare variants
Rare variants are replaced by the most frequent allele. This method is used to exclude rare variants in some analyses as in McDonald-Kreitman Test (McDonald & Kreitman, 1991, Nature 351 pp652-654). For an application, see for example (Fay et al. 2001, Genetics 158 pp 1227-1234).
- Parameters
-
site a Site gCode The genetic code according to which stop codons are specified. freqmin a double, allele in frequency stricly lower than freqmin are replaced
- Exceptions
-
AlphabetException If the alphabet associated to the site is not a codon alphabet. EmptySiteException If the site has size 0.
Definition at line 158 of file CodonSiteTools.cpp.
References bpp::CodonAlphabet::getFirstPosition(), bpp::SymbolListTools::getFrequencies(), bpp::CodonAlphabet::getNucleicAlphabet(), bpp::CodonAlphabet::getSecondPosition(), bpp::CodonAlphabet::getThirdPosition(), bpp::BasicSymbolList::getValue(), bpp::AlphabetTools::isCodonAlphabet(), and bpp::SiteTools::isConstant().
Referenced by numberOfSubsitutions().
◆ getCounts() [1/4]
|
inlinestaticinherited |
Count all states in the list.
- Parameters
-
list The list. counts The output map to store the counts (existing counts will be incremented).
Definition at line 70 of file SymbolListTools.h.
References bpp::SymbolList::getContent().
Referenced by bpp::SiteTools::getNumberOfDistinctCharacters(), bpp::SequenceApplicationTools::getSitesToAnalyse(), bpp::SiteTools::isParsimonyInformativeSite(), and numberOfNonSynonymousSubstitutions().
◆ getCounts() [2/4]
|
inlinestaticinherited | ||||||||||||||||||||||||
Count all pair of states for two lists of the same size.
NB: The two lists do node need to share the same alphabet! The states of the first list will be used as the first index in the output, and the ones from the second list as the second index.
- Parameters
-
list1 The first list. list2 The second list. counts The output map to store the counts (existing counts will be incremented).
Definition at line 90 of file SymbolListTools.h.
◆ getCounts() [3/4]
|
staticinherited |
Count all states in the list, optionaly resolving unknown characters.
For instance, in DNA, N will be counted as A=1/4,T=1/4,C=1/4,G=1/4.
- Parameters
-
list The list. counts The output map to store the counts (existing ocunts will be incremented). resolveUnknowns Tell is unknown characters must be resolved. For instance, in DNA, N will be counted as A=1/4,T=1/4,C=1/4,G=1/4.
- Returns
- A map with all states and corresponding counts.
Definition at line 51 of file SymbolListTools.cpp.
References bpp::Alphabet::getAlias(), bpp::SymbolList::getAlphabet(), and bpp::SymbolList::getContent().
◆ getCounts() [4/4]
|
staticinherited | ||||||||||||||||||||||||||||
Count all pair of states for two lists of the same size, optionaly resolving unknown characters.
For instance, in DNA, N will be counted as A=1/4,T=1/4,C=1/4,G=1/4.
NB: The two lists do node need to share the same alphabet! The states of the first list will be used as the first index in the output, and the ones from the second list as the second index.
- Parameters
-
list1 The first list. list2 The second list. counts The output map to store the counts (existing ocunts will be incremented). resolveUnknowns Tell is unknown characters must be resolved. For instance, in DNA, N will be counted as A=1/4,T=1/4,C=1/4,G=1/4.
- Returns
- A map with all states and corresponding counts.
Definition at line 73 of file SymbolListTools.cpp.
◆ getFrequencies() [1/2]
|
staticinherited |
Get all states frequencies in the list.
- Parameters
-
list The list. resolveUnknowns Tell is unknown characters must be resolved. For instance, in DNA, N will be counted as A=1/4,T=1/4,C=1/4,G=1/4. frequencies The output map with all states and corresponding frequencies. Existing frequencies will be erased if any.
Definition at line 96 of file SymbolListTools.cpp.
References bpp::SymbolList::size().
Referenced by generateCodonSiteWithoutRareVariant(), bpp::SiteContainerTools::getConsensus(), bpp::SequenceApplicationTools::getSitesToAnalyse(), meanNumberOfSynonymousPositions(), piNonSynonymous(), piSynonymous(), and bpp::SiteContainerTools::removeGapSites().
◆ getFrequencies() [2/2]
|
staticinherited | ||||||||||||||||||||||||||||
Get all state pairs frequencies for two lists of the same size..
- Parameters
-
list1 The first list. list2 The second list. resolveUnknowns Tell is unknown characters must be resolved. For instance, in DNA, N will be counted as A=1/4,T=1/4,C=1/4,G=1/4. frequencies The output map with all state pairs and corresponding frequencies. Existing frequencies will be erased if any.
Definition at line 107 of file SymbolListTools.cpp.
◆ getGCContent()
|
staticinherited | ||||||||||||||||||||||||
Get the GC content of a symbol list.
- Parameters
-
list The list.
- Returns
- The proportion of G and C states in the list.
- Parameters
-
ignoreUnresolved Do not count unresolved states. Otherwise, weight by each state probability in case of ambiguity (e.g. the R state counts for 0.5). ignoreGap Do not count gaps in total.
- Exceptions
-
AlphabetException If the list is not made of nucleotide states.
Definition at line 119 of file SymbolListTools.cpp.
◆ getNumberOfDistinctPositions()
|
staticinherited | ||||||||||||||||||||
Get the number of distinct positions.
The comparison in achieved from position 0 to the minimum size of the two vectors.
- Parameters
-
l1 SymbolList 1. l2 SymbolList 2.
- Returns
- The number of distinct positions.
- Exceptions
-
AlphabetMismatchException if the two lists have not the same alphabet type.
Definition at line 158 of file SymbolListTools.cpp.
◆ getNumberOfPositionsWithoutGap()
|
staticinherited | ||||||||||||||||||||
Get the number of positions without gap.
The comparison in achieved from position 0 to the minimum size of the two vectors.
- Parameters
-
l1 SymbolList 1. l2 SymbolList 2.
- Returns
- The number of positions without gap.
- Exceptions
-
AlphabetMismatchException if the two lists have not the same alphabet type.
Definition at line 169 of file SymbolListTools.cpp.
◆ hasGapOrStop()
|
static | ||||||||||||||||||||
Method to know if a codon site contains gap(s) or stop codons.
- Parameters
-
site a Site gCode The genetic code according to which stop codons are specified.
- Exceptions
-
AlphabetException If the alphabet associated to the site is not a codon alphabet.
Definition at line 60 of file CodonSiteTools.cpp.
References bpp::AlphabetTools::isCodonAlphabet().
◆ hasStop()
|
static | ||||||||||||||||||||
Method to know if a codon site contains stop codon or not.
- Parameters
-
site a Site gCode The genetic code according to which stop codons are specified.
- Exceptions
-
AlphabetException If the alphabet associated to the site is not a codon alphabet.
Definition at line 75 of file CodonSiteTools.cpp.
References bpp::AlphabetTools::isCodonAlphabet().
Referenced by bpp::SiteContainerTools::removeStopCodonSites().
◆ isFourFoldDegenerated()
|
static |
- Returns
- True if all sequences have a fourfold degenerated codon in the site (that is, if a mutation in the fourth position does not change the aminoacid).
- Parameters
-
site The site to analyze. gCode The genetic code to use.
If non-synonymous mutation
Definition at line 807 of file CodonSiteTools.cpp.
References bpp::BasicSymbolList::getValue(), bpp::SiteTools::isConstant(), bpp::GeneticCode::isFourFoldDegenerated(), isSynonymousPolymorphic(), and bpp::BasicSymbolList::size().
◆ isMonoSitePolymorphic()
|
static | ||||||||||||||||||
Method to know if a polymorphic codon site is polymorphic at only one site.
- Parameters
-
site a Site
- Exceptions
-
AlphabetException If the alphabet associated to the site is not a codon alphabet. EmptySiteException If the site has size 0.
Definition at line 90 of file CodonSiteTools.cpp.
References bpp::CodonAlphabet::getFirstPosition(), bpp::CodonAlphabet::getNucleicAlphabet(), bpp::CodonAlphabet::getSecondPosition(), bpp::CodonAlphabet::getThirdPosition(), bpp::AlphabetTools::isCodonAlphabet(), and bpp::SiteTools::isConstant().
◆ isSynonymousPolymorphic()
|
static | ||||||||||||||||||||||||||||
Method to know if polymorphism at a codon site is synonymous.
- Parameters
-
site a Site gCode a GeneticCode
- Exceptions
-
AlphabetException If the alphabet associated to the site is not a codon alphabet. AlphabetMismatchException If the codon alphabet of the site do not match the codon alphabet of the genetic code. EmptySiteException If the site has size 0.
Definition at line 128 of file CodonSiteTools.cpp.
References bpp::AlphabetTools::isCodonAlphabet(), and bpp::SiteTools::isConstant().
Referenced by isFourFoldDegenerated().
◆ meanNumberOfSynonymousPositions()
|
static | ||||||||||||||||||||||||||||||||
Return the mean number of synonymous positions per codon site.
A site is consider as x% synonymous if x% of the possible mutations are synonymous Transition/transversion ratio can be taken into account (use the variable ratio) The mean is computed over the VectorSite.
Unresolved and stop codons are counted as 0.
- Parameters
-
site a Site gCode a GeneticCode ratio a double Set by default to 1
- Exceptions
-
AlphabetException If the alphabet associated to the site is not a codon alphabet. AlphabetMismatchException If the codon alphabet of the site do not match the codon alphabet of the genetic code. EmptySiteException If the site has size 0.
Definition at line 563 of file CodonSiteTools.cpp.
References bpp::SymbolListTools::getFrequencies(), and bpp::AlphabetTools::isCodonAlphabet().
◆ numberOfDifferences()
|
static |
Compute the number of differences between two codons.
- Parameters
-
i a int j a int ca a CodonAlphabet
Definition at line 220 of file CodonSiteTools.cpp.
References bpp::CodonAlphabet::getFirstPosition(), bpp::CodonAlphabet::getSecondPosition(), and bpp::CodonAlphabet::getThirdPosition().
◆ numberOfNonSynonymousSubstitutions()
|
static | ||||||||||||||||||||||||||||||||
Return the number of Non Synonymous subsitutions per codon site.
It is assumed that the path linking amino acids only involved one substitution by step.
Rare variants (<= freqmin) can be excluded. In case of complex codon, the path that gives the minimum number of non-synonymous changes is chosen. The argument minchange=true is sent to numberOfSynonymousDifferences used in this method. Otherwise, a non-integer number could be return.
- Parameters
-
site a Site gCode a GeneticCode freqmin a double To exclude snp in frequency strictly lower than freqmin (by default freqmin = 0).
- Exceptions
-
AlphabetException If the alphabet associated to the site is not a codon alphabet. AlphabetMismatchException If the codon alphabet of the site do not match the codon alphabet of the genetic code. EmptySiteException If the site has size 0.
Definition at line 633 of file CodonSiteTools.cpp.
References bpp::SymbolListTools::getCounts(), bpp::SiteTools::hasGap(), bpp::AlphabetTools::isCodonAlphabet(), and bpp::SiteTools::isConstant().
◆ numberOfSubsitutions()
|
static | ||||||||||||||||||||||||||||
Return the number of subsitutions per codon site.
No recombination is assumed, that is in complex codon homoplasy is assumed. Example:
Here, 3 substitutions are counted. Assuming that the last codon (AGC) is a recombinant between ATC and AGT would have lead to counting only 2 subsitutions.
Rare variants (<= freqmin) can be excluded.
- Parameters
-
site a Site gCode a GeneticCode freqmin a double To exclude snp in frequency strictly lower than freqmin (by default freqmin = 0)
- Exceptions
-
AlphabetException If the alphabet associated to the site is not a codon alphabet. EmptySiteException If the site has size 0.
Definition at line 588 of file CodonSiteTools.cpp.
References generateCodonSiteWithoutRareVariant(), bpp::CodonAlphabet::getFirstPosition(), bpp::CodonAlphabet::getNucleicAlphabet(), bpp::SiteTools::getNumberOfDistinctCharacters(), bpp::CodonAlphabet::getSecondPosition(), bpp::CodonAlphabet::getThirdPosition(), bpp::BasicSymbolList::getValue(), bpp::SiteTools::hasGap(), bpp::AlphabetTools::isCodonAlphabet(), bpp::SiteTools::isConstant(), and bpp::BasicSymbolList::size().
◆ numberOfSynonymousDifferences()
|
static |
Compute the number of synonymous differences between two codons.
For complex codon: If minchange = false (default option) the different paths are equally weighted. If minchange = true the path with the minimum number of non-synonymous change is chosen. Paths included stop codons are excluded.
- Parameters
-
i a int j a int gCode a GeneticCode minchange a boolean set by default to false
Definition at line 234 of file CodonSiteTools.cpp.
References bpp::GeneticCode::areSynonymous(), bpp::CodonAlphabet::getCodon(), bpp::WordAlphabet::getPositions(), bpp::GeneticCode::getSourceAlphabet(), and bpp::GeneticCode::isStop().
◆ numberOfSynonymousPositions()
|
static | ||||||||||||||||||||||||
Return the number of synonymous positions of a codon.
A site is consider as x% synonymous if x% of the possible mutations are synonymous Transition/transversion ratio can be taken into account (use the variable ratio)
Unresolved codons and stop codon will return a value of 0.
- Parameters
-
i a int gCode a GeneticCode ratio a double set by default to 1
Definition at line 522 of file CodonSiteTools.cpp.
References bpp::CodonAlphabet::getCodon(), bpp::WordAlphabet::getPositions(), and bpp::WordAlphabet::isUnresolved().
◆ piNonSynonymous()
|
static | ||||||||||||||||||||||||||||||||
Compute the non-synonymous pi per codon site.
The following formula is used:
![\[ pi = frac{n}{n-1}\sum_{i,j}x_{i}x_{j}P_{ij} \]](form_6.png)
where n is the number of sequence, 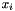 and
and 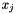 the frequencies of each codon type occuring at the site
the frequencies of each codon type occuring at the site 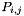 the number of nonsynonymous difference between these codons. Be careful: here, pi is not normalized by the number of non-synonymous sites. If minchange = false (default option) the different paths are equally weighted. If minchange = true the path with the minimum number of non-synonymous change is chosen.
the number of nonsynonymous difference between these codons. Be careful: here, pi is not normalized by the number of non-synonymous sites. If minchange = false (default option) the different paths are equally weighted. If minchange = true the path with the minimum number of non-synonymous change is chosen.
- Parameters
-
site a Site gCode a GeneticCode minchange a boolean set by default to false
- Exceptions
-
AlphabetException If the alphabet associated to the site is not a codon alphabet. AlphabetMismatchException If the codon alphabet of the site do not match the codon alphabet of the genetic code. EmptySiteException If the site has size 0.
Definition at line 485 of file CodonSiteTools.cpp.
References bpp::SymbolListTools::getFrequencies(), bpp::AlphabetTools::isCodonAlphabet(), and bpp::SiteTools::isConstant().
◆ piSynonymous()
|
static | ||||||||||||||||||||||||||||||||
Compute the synonymous pi per codon site.
The following formula is used:
where n is the number of sequence, and the frequencies of each codon type occuring at the site the number of synonymous difference between these codons. Be careful: here, pi is not normalized by the number of synonymous sites.
If minchange = false (default option) the different paths are equally weighted. If minchange = true the path with the minimum number of non-synonymous change is chosen.
- Parameters
-
site a Site gCode a GeneticCode minchange a boolean set by default to false
- Exceptions
-
AlphabetException If the alphabet associated to the site is not a codon alphabet. AlphabetMismatchException If the codon alphabet of the site do not match the codon alphabet of the genetic code. EmptySiteException If the site has size 0.
Definition at line 453 of file CodonSiteTools.cpp.
References bpp::SymbolListTools::getFrequencies(), bpp::AlphabetTools::isCodonAlphabet(), and bpp::SiteTools::isConstant().
The documentation for this class was generated from the following files:
- Bpp/Seq/CodonSiteTools.h
- Bpp/Seq/CodonSiteTools.cpp
