create_atoms command
Syntax
create_atoms type style args keyword values ...
type = atom type (1-Ntypes) of atoms to create (offset for molecule creation)
style = box or region or single or random
box args = none region args = region-ID region-ID = particles will only be created if contained in the region single args = x y z x,y,z = coordinates of a single particle (distance units) random args = N seed region-ID N = number of particles to create seed = random # seed (positive integer) region-ID = create atoms within this region, use NULL for entire simulation box
zero or more keyword/value pairs may be appended
keyword = mol or basis or remap or var or set or units
mol value = template-ID seed template-ID = ID of molecule template specified in a separate molecule command seed = random # seed (positive integer) basis values = M itype M = which basis atom itype = atom type (1-N) to assign to this basis atom remap value = yes or no var value = name = variable name to evaluate for test of atom creation set values = dim name dim = x or y or z name = name of variable to set with x, y, or z atom position rotate values = Rx Ry Rz theta Rx,Ry,Rz = rotation vector for single molecule theta = rotation angle for single molecule (degrees) units value = lattice or box lattice = the geometry is defined in lattice units box = the geometry is defined in simulation box units
Examples
create_atoms 1 box
create_atoms 3 region regsphere basis 2 3
create_atoms 3 single 0 0 5
create_atoms 1 box var v set x xpos set y ypos
Description
This command creates atoms (or molecules) on a lattice, or a single atom (or molecule), or a random collection of atoms (or molecules), as an alternative to reading in their coordinates explicitly via a read_data or read_restart command. A simulation box must already exist, which is typically created via the create_box command. Before using this command, a lattice must also be defined using the lattice command, unless you specify the single style with units = box or the random style. For the remainder of this doc page, a created atom or molecule is referred to as a “particle”.
If created particles are individual atoms, they are assigned the specified atom type, though this can be altered via the basis keyword as discussed below. If molecules are being created, the type of each atom in the created molecule is specified in the file read by the molecule command, and those values are added to the specified atom type. E.g. if type = 2, and the file specifies atom types 1,2,3, then each created molecule will have atom types 3,4,5.
For the box style, the create_atoms command fills the entire simulation box with particles on the lattice. If your simulation box is periodic, you should insure its size is a multiple of the lattice spacings, to avoid unwanted atom overlaps at the box boundaries. If your box is periodic and a multiple of the lattice spacing in a particular dimension, LAMMPS is careful to put exactly one particle at the boundary (on either side of the box), not zero or two.
For the region style, a geometric volume is filled with particles on the lattice. This volume what is inside the simulation box and is also consistent with the region volume. See the region command for details. Note that a region can be specified so that its “volume” is either inside or outside a geometric boundary. Also note that if your region is the same size as a periodic simulation box (in some dimension), LAMMPS does not implement the same logic described above as for the box style, to insure exactly one particle at periodic boundaries. if this is what you desire, you should either use the box style, or tweak the region size to get precisely the particles you want.
For the single style, a single particle is added to the system at the specified coordinates. This can be useful for debugging purposes or to create a tiny system with a handful of particles at specified positions.
For the random style, N particles are added to the system at randomly generated coordinates, which can be useful for generating an amorphous system. The particles are created one by one using the speficied random number seed, resulting in the same set of particles coordinates, independent of how many processors are being used in the simulation. If the region-ID argument is specified as NULL, then the created particles will be anywhere in the simulation box. If a region-ID is specified, a geometric volume is filled which is both inside the simulation box and is also consistent with the region volume. See the region command for details. Note that a region can be specified so that its “volume” is either inside or outside a geometric boundary.
Note
Particles generated by the random style will typically be highly overlapped which will cause many interatomic potentials to compute large energies and forces. Thus you should either perform an energy minimization or run dynamics with fix nve/limit to equilibrate such a system, before running normal dynamics.
Note that this command adds particles to those that already exist. This means it can be used to add particles to a system previously read in from a data or restart file. Or the create_atoms command can be used multiple times, to add multiple sets of particles to the simulation. For example, grain boundaries can be created, by interleaving create_atoms with lattice commands specifying different orientations. By using the create_atoms command in conjunction with the delete_atoms command, reasonably complex geometries can be created, or a protein can be solvated with a surrounding box of water molecules.
In all these cases, care should be taken to insure that new atoms do not overlap existing atoms inappropriately, especially if molecules are being added. The delete_atoms command can be used to remove overlapping atoms or molecules.
Individual atoms are inserted by this command, unless the mol keyword is used. It specifies a template-ID previously defined using the molecule command, which reads a file that defines the molecule. The coordinates, atom types, charges, etc, as well as any bond/angle/etc and special neighbor information for the molecule can be specified in the molecule file. See the molecule command for details. The only settings required to be in this file are the coordinates and types of atoms in the molecule.
Using a lattice to add molecules, e.g. via the box or region or single styles, is exactly the same as adding atoms on lattice points, except that entire molecules are added at each point, i.e. on the point defined by each basis atom in the unit cell as it tiles the simulation box or region. This is done by placing the geometric center of the molecule at the lattice point, and giving the molecule a random orientation about the point. The random seed specified with the mol keyword is used for this operation, and the random numbers generated by each processor are different. This means the coordinates of individual atoms (in the molecules) will be different when running on different numbers of processors, unlike when atoms are being created in parallel.
Also note that because of the random rotations, it may be important to use a lattice with a large enough spacing that adjacent molecules will not overlap, regardless of their relative orientations.
Note
If the create_box command is used to create the simulation box, followed by the create_atoms command with its mol option for adding molecules, then you typically need to use the optional keywords allowed by the create_box command for extra bonds (angles,etc) or extra special neighbors. This is because by default, the create_box command sets up a non-molecular system which doesn’t allow molecules to be added.
This is the meaning of the other allowed keywords.
The basis keyword is only used when atoms (not molecules) are being created. It specifies an atom type that will be assigned to specific basis atoms as they are created. See the lattice command for specifics on how basis atoms are defined for the unit cell of the lattice. By default, all created atoms are assigned the argument type as their atom type.
The remap keyword only applies to the single style. If it is set to yes, then if the specified position is outside the simulation box, it will mapped back into the box, assuming the relevant dimensions are periodic. If it is set to no, no remapping is done and no particle is created if its position is outside the box.
The var and set keywords can be used together to provide a criterion for accepting or rejecting the addition of an individual atom, based on its coordinates. The name specified for the var keyword is the name of an equal-style variable which should evaluate to a zero or non-zero value based on one or two or three variables which will store the x, y, or z coordinates of an atom (one variable per coordinate). If used, these other variables must be internal-style variables defined in the input script; their initial numeric value can be anything. They must be internal-style variables, because this command resets their values directly. The set keyword is used to identify the names of these other variables, one variable for the x-coordinate of a created atom, one for y, and one for z.
When an atom is created, its x,y,z coordinates become the values for any set variable that is defined. The var variable is then evaluated. If the returned value is 0.0, the atom is not created. If it is non-zero, the atom is created.
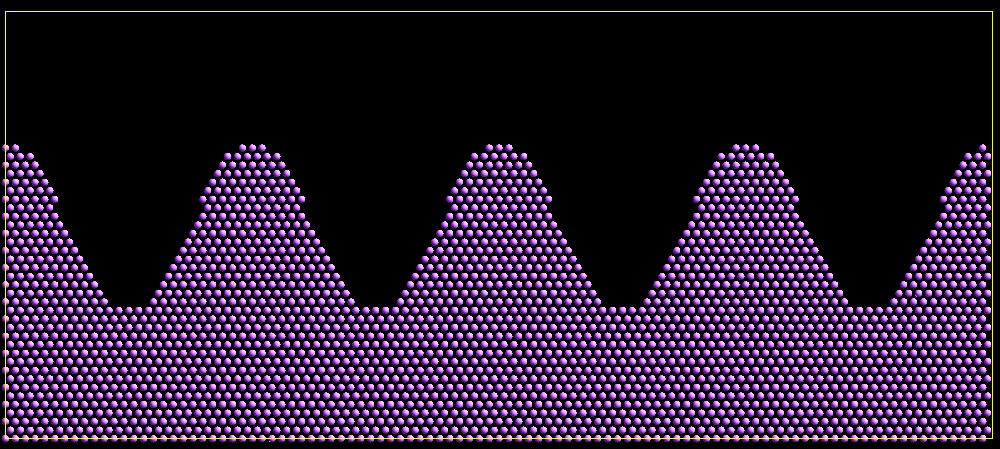
As an example, these commands can be used in a 2d simulation, to create a sinusoidal surface. Note that the surface is “rough” due to individual lattice points being “above” or “below” the mathematical expression for the sinusoidal curve. If a finer lattice were used, the sinusoid would appear to be “smoother”. Also note the use of the “xlat” and “ylat” thermo_style keywords which converts lattice spacings to distance. Click on the image for a larger version.
variable x equal 100 variable y equal 25 lattice hex 0.8442 region box block 0 $x 0 $y -0.5 0.5 create_box 1 box variable xx equal 0.0 variable yy equal 0.0 variable v equal "(0.2*v_y*ylat * cos(v_xx/xlat * 2.0*PI*4.0/v_x) + 0.5*v_y*ylat - v_yy) > 0.0" create_atoms 1 box var v set x xx set y yy
The rotate keyword can be used with the single style, when adding a single molecule to specify the orientation at which the molecule is inserted. The axis of rotation is determined by the rotation vector (Rx,Ry,Rz) that goes through the insertion point. The specified theta determines the angle of rotation around that axis. Note that the direction of rotation for the atoms around the rotation axis is consistent with the right-hand rule: if your right-hand’s thumb points along R, then your fingers wrap around the axis in the direction of rotation.
The units keyword determines the meaning of the distance units used to specify the coordinates of the one particle created by the single style. A box value selects standard distance units as defined by the units command, e.g. Angstroms for units = real or metal. A lattice value means the distance units are in lattice spacings.
Atom IDs are assigned to created atoms in the following way. The collection of created atoms are assigned consecutive IDs that start immediately following the largest atom ID existing before the create_atoms command was invoked. When a simulation is performed on different numbers of processors, there is no guarantee a particular created atom will be assigned the same ID. If molecules are being created, molecule IDs are assigned to created molecules in a similar fashion.
Aside from their ID, atom type, and xyz position, other properties of created atoms are set to default values, depending on which quantities are defined by the chosen atom style. See the atom style command for more details. See the set and velocity commands for info on how to change these values.
- charge = 0.0
- dipole moment magnitude = 0.0
- diameter = 1.0
- shape = 0.0 0.0 0.0
- density = 1.0
- volume = 1.0
- velocity = 0.0 0.0 0.0
- angular velocity = 0.0 0.0 0.0
- angular momentum = 0.0 0.0 0.0
- quaternion = (1,0,0,0)
- bonds, angles, dihedrals, impropers = none
If molecules are being created, these defaults can be overridden by values specified in the file read by the molecule command. E.g. the file typically defines bonds (angles,etc) between atoms in the molecule, and can optionally define charges on each atom.
Note that the sphere atom style sets the default particle diameter to 1.0 as well as the density. This means the mass for the particle is not 1.0, but is PI/6 * diameter^3 = 0.5236.
Note that the ellipsoid atom style sets the default particle shape to (0.0 0.0 0.0) and the density to 1.0 which means it is a point particle, not an ellipsoid, and has a mass of 1.0.
Note that the peri style sets the default volume and density to 1.0 and thus also set the mass for the particle to 1.0.
The set command can be used to override many of these default settings.
Restrictions
An atom_style must be previously defined to use this command.
A rotation vector specified for a single molecule must be in the z-direction for a 2d model.
Default
The default for the basis keyword is that all created atoms are assigned the argument type as their atom type (when single atoms are being created). The other defaults are remap = no, rotate = random, and units = lattice.