fix shake command
fix rattle command
Syntax
fix ID group-ID style tol iter N constraint values ... keyword value ...
ID, group-ID are documented in fix command
style = shake or rattle = style name of this fix command
tol = accuracy tolerance of SHAKE solution
iter = max # of iterations in each SHAKE solution
N = print SHAKE statistics every this many timesteps (0 = never)
one or more constraint/value pairs are appended
constraint = b or a or t or m
b values = one or more bond types a values = one or more angle types t values = one or more atom types m value = one or more mass values
zero or more keyword/value pairs may be appended
keyword = mol
mol value = template-ID template-ID = ID of molecule template specified in a separate molecule command
Examples
fix 1 sub shake 0.0001 20 10 b 4 19 a 3 5 2
fix 1 sub shake 0.0001 20 10 t 5 6 m 1.0 a 31
fix 1 sub shake 0.0001 20 10 t 5 6 m 1.0 a 31 mol myMol
fix 1 sub rattle 0.0001 20 10 t 5 6 m 1.0 a 31
fix 1 sub rattle 0.0001 20 10 t 5 6 m 1.0 a 31 mol myMol
Description
Apply bond and angle constraints to specified bonds and angles in the simulation by either the SHAKE or RATTLE algorithms. This typically enables a longer timestep.
SHAKE vs RATTLE:
The SHAKE algorithm was invented for schemes such as standard Verlet timesteppnig, where only the coordinates are integrated and the velocities are approximated as finite differences to the trajectories (Ryckaert et al. (1977)). If the velocities are integrated explicitly, as with velocity Verlet which is what LAMMPS uses as an integration method, a second set of constraining forces is required in order to eliminate velocity components along the bonds (Andersen (1983)).
In order to formulate individual constraints for SHAKE and RATTLE, focus on a single molecule whose bonds are constrained. Let Ri and Vi be the position and velocity of atom i at time n, for i=1,...,N, where N is the number of sites of our reference molecule. The distance vector between sites i and j is given by
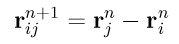
The constraints can then be formulated as
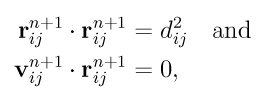
The SHAKE algorithm satisfies the first condition, i.e. the sites at time n+1 will have the desired separations Dij immediately after the coordinates are integrated. If we also enforce the second condition, the velocity components along the bonds will vanish. RATTLE satisfies both conditions. As implemented in LAMMPS, fix rattle uses fix shake for satisfying the coordinate constraints. Therefore the settings and optional keywords are the same for both fixes, and all the information below about SHAKE is also relevant for RATTLE.
SHAKE:
Each timestep the specified bonds and angles are reset to their equilibrium lengths and angular values via the SHAKE algorithm (Ryckaert et al. (1977)). This is done by applying an additional constraint force so that the new positions preserve the desired atom separations. The equations for the additional force are solved via an iterative method that typically converges to an accurate solution in a few iterations. The desired tolerance (e.g. 1.0e-4 = 1 part in 10000) and maximum # of iterations are specified as arguments. Setting the N argument will print statistics to the screen and log file about regarding the lengths of bonds and angles that are being constrained. Small delta values mean SHAKE is doing a good job.
In LAMMPS, only small clusters of atoms can be constrained. This is so the constraint calculation for a cluster can be performed by a single processor, to enable good parallel performance. A cluster is defined as a central atom connected to others in the cluster by constrained bonds. LAMMPS allows for the following kinds of clusters to be constrained: one central atom bonded to 1 or 2 or 3 atoms, or one central atom bonded to 2 others and the angle between the 3 atoms also constrained. This means water molecules or CH2 or CH3 groups may be constrained, but not all the C-C backbone bonds of a long polymer chain.
The b constraint lists bond types that will be constrained. The t constraint lists atom types. All bonds connected to an atom of the specified type will be constrained. The m constraint lists atom masses. All bonds connected to atoms of the specified masses will be constrained (within a fudge factor of MASSDELTA specified in fix_shake.cpp). The a constraint lists angle types. If both bonds in the angle are constrained then the angle will also be constrained if its type is in the list.
For all constraints, a particular bond is only constrained if both atoms in the bond are in the group specified with the SHAKE fix.
The degrees-of-freedom removed by SHAKE bonds and angles are accounted for in temperature and pressure computations. Similarly, the SHAKE contribution to the pressure of the system (virial) is also accounted for.
Note
This command works by using the current forces on atoms to caculate an additional constraint force which when added will leave the atoms in positions that satisfy the SHAKE constraints (e.g. bond length) after the next time integration step. If you define fixes (e.g. fix efield) that add additional force to the atoms after fix shake operates, then this fix will not take them into account and the time integration will typically not satisfy the SHAKE constraints. The solution for this is to make sure that fix shake is defined in your input script after any other fixes which add or change forces (to atoms that fix shake operates on).
The mol keyword should be used when other commands, such as fix deposit or fix pour, add molecules on-the-fly during a simulation, and you wish to contrain the new molecules via SHAKE. You specify a template-ID previously defined using the molecule command, which reads a file that defines the molecule. You must use the same template-ID that the command adding molecules uses. The coordinates, atom types, special bond restrictions, and SHAKE info can be specified in the molecule file. See the molecule command for details. The only settings required to be in this file (by this command) are the SHAKE info of atoms in the molecule.
Styles with a suffix are functionally the same as the corresponding style without the suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section 5 of the manual. The accelerated styles take the same arguments and should produce the same results, except for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages, respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
RATTLE:
The velocity constraints lead to a linear system of equations which can be solved analytically. The implementation of the algorithm in LAMMPS closely follows (Andersen (1983)).
Note
The fix rattle command modifies forces and velocities and thus should be defined after all other integration fixes in your input script. If you define other fixes that modify velocities or forces after fix rattle operates, then fix rattle will not take them into account and the overall time integration will typically not satisfy the RATTLE constraints. You can check whether the constraints work correctly by setting the value of RATTLE_DEBUG in src/fix_rattle.cpp to 1 and recompiling LAMMPS.
Restart, fix_modify, output, run start/stop, minimize info:
No information about these fixes is written to binary restart files. None of the fix_modify options are relevant to these fixes. No global or per-atom quantities are stored by these fixes for access by various output commands. No parameter of these fixes can be used with the start/stop keywords of the run command. These fixes are not invoked during energy minimization.
Restrictions
These fixes are part of the RIGID package. They are only enabled if LAMMPS was built with that package. See the Making LAMMPS section for more info.
For computational efficiency, there can only be one shake or rattle fix defined in a simulation.
If you use a tolerance that is too large or a max-iteration count that is too small, the constraints will not be enforced very strongly, which can lead to poor energy conservation. You can test for this in your system by running a constant NVE simulation with a particular set of SHAKE parameters and monitoring the energy versus time.
SHAKE or RATTLE should not be used to contrain an angle at 180 degrees (e.g. linear CO2 molecule). This causes numeric difficulties.
Related commands: none
Default: none
(Ryckaert) J.-P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J of Comp Phys, 23, 327-341 (1977).
(Andersen) H. Andersen, J of Comp Phys, 52, 24-34 (1983).