pair_style adp command
pair_style adp/omp command
Syntax
pair_style adp
Examples
pair_style adp pair_coeff * * Ta.adp Ta pair_coeff * * ../potentials/AlCu.adp Al Al Cu
Description
Style adp computes pairwise interactions for metals and metal alloys using the angular dependent potential (ADP) of (Mishin), which is a generalization of the embedded atom method (EAM) potential. The LAMMPS implementation is discussed in (Singh). The total energy Ei of an atom I is given by
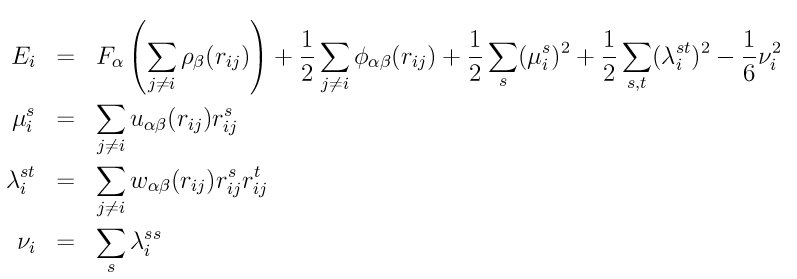
where F is the embedding energy which is a function of the atomic electron density rho, phi is a pair potential interaction, alpha and beta are the element types of atoms I and J, and s and t = 1,2,3 and refer to the cartesian coordinates. The mu and lambda terms represent the dipole and quadruple distortions of the local atomic environment which extend the original EAM framework by introducing angular forces.
Note that unlike for other potentials, cutoffs for ADP potentials are not set in the pair_style or pair_coeff command; they are specified in the ADP potential files themselves. Likewise, the ADP potential files list atomic masses; thus you do not need to use the mass command to specify them.
The NIST WWW site distributes and documents ADP potentials:
http://www.ctcms.nist.gov/potentials
Note that these must be converted into the extended DYNAMO setfl format discussed below.
The NIST site is maintained by Chandler Becker (cbecker at nist.gov) who is good resource for info on interatomic potentials and file formats.
Only a single pair_coeff command is used with the adp style which specifies an extended DYNAMO setfl file, which contains information for M elements. These are mapped to LAMMPS atom types by specifying N additional arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom types:
- filename
- N element names = mapping of extended setfl elements to atom types
See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example, the potentials/AlCu.adp file, included in the potentials directory of the LAMMPS distrbution, is an extended setfl file which has tabulated ADP values for w elements and their alloy interactions: Cu and Al. If your LAMMPS simulation has 4 atoms types and you want the 1st 3 to be Al, and the 4th to be Cu, you would use the following pair_coeff command:
pair_coeff * * AlCu.adp Al Al Al Cu
The 1st 2 arguments must be * * so as to span all LAMMPS atom types. The first three Al arguments map LAMMPS atom types 1,2,3 to the Al element in the extended setfl file. The final Cu argument maps LAMMPS atom type 4 to the Al element in the extended setfl file. Note that there is no requirement that your simulation use all the elements specified by the extended setfl file.
If a mapping value is specified as NULL, the mapping is not performed. This can be used when an adp potential is used as part of the hybrid pair style. The NULL values are placeholders for atom types that will be used with other potentials.
Adp files in the potentials directory of the LAMMPS distribution have an ”.adp” suffix. A DYNAMO setfl file extended for ADP is formatted as follows. Basically it is the standard setfl format with additional tabulated functions u and w added to the file after the tabulated pair potentials. See the pair_eam command for further details on the setfl format.
- lines 1,2,3 = comments (ignored)
- line 4: Nelements Element1 Element2 ... ElementN
- line 5: Nrho, drho, Nr, dr, cutoff
Following the 5 header lines are Nelements sections, one for each element, each with the following format:
- line 1 = atomic number, mass, lattice constant, lattice type (e.g. FCC)
- embedding function F(rho) (Nrho values)
- density function rho(r) (Nr values)
Following the Nelements sections, Nr values for each pair potential phi(r) array are listed for all i,j element pairs in the same format as other arrays. Since these interactions are symmetric (i,j = j,i) only phi arrays with i >= j are listed, in the following order: i,j = (1,1), (2,1), (2,2), (3,1), (3,2), (3,3), (4,1), ..., (Nelements, Nelements). The tabulated values for each phi function are listed as r*phi (in units of eV-Angstroms), since they are for atom pairs, the same as for other EAM files.
After the phi(r) arrays, each of the u(r) arrays are listed in the same order with the same assumptions of symmetry. Directly following the u(r), the w(r) arrays are listed. Note that phi(r) is the only array tabulated with a scaling by r.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section 5 of the manual. The accelerated styles take the same arguments and should produce the same results, except for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages, respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, where types I and J correspond to two different element types, no special mixing rules are needed, since the ADP potential files specify alloy interactions explicitly.
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in tabulated potential files. Thus, you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the inner, middle, outer keywords.
Restrictions
This pair style is part of the MANYBODY package. It is only enabled if LAMMPS was built with that package (which it is by default).