pair_style resquared command
pair_style resquared/gpu command
pair_style resquared/omp command
Syntax
pair_style resquared cutoff
- cutoff = global cutoff for interactions (distance units)
Examples
pair_style resquared 10.0 pair_coeff * * 1.0 1.0 1.7 3.4 3.4 1.0 1.0 1.0
Description
Style resquared computes the RE-squared anisotropic interaction (Everaers), (Babadi) between pairs of ellipsoidal and/or spherical Lennard-Jones particles. For ellipsoidal interactions, the potential considers the ellipsoid as being comprised of small spheres of size sigma. LJ particles are a single sphere of size sigma. The distinction is made to allow the pair style to make efficient calculations of ellipsoid/solvent interactions.
Details for the equations used are given in the references below and in this supplementary document.
Use of this pair style requires the NVE, NVT, or NPT fixes with the asphere extension (e.g. fix nve/asphere) in order to integrate particle rotation. Additionally, atom_style ellipsoid should be used since it defines the rotational state and the size and shape of each ellipsoidal particle.
The following coefficients must be defined for each pair of atoms types via the pair_coeff command as in the examples above, or in the data file or restart files read by the read_data or read_restart commands:
- A12 = Energy Prefactor/Hamaker constant (energy units)
- sigma = atomic interaction diameter (distance units)
- epsilon_i_a = relative well depth of type I for side-to-side interactions
- epsilon_i_b = relative well depth of type I for face-to-face interactions
- epsilon_i_c = relative well depth of type I for end-to-end interactions
- epsilon_j_a = relative well depth of type J for side-to-side interactions
- epsilon_j_b = relative well depth of type J for face-to-face interactions
- epsilon_j_c = relative well depth of type J for end-to-end interactions
- cutoff (distance units)
The parameters used depend on the type of the interacting particles, i.e. ellipsoids or LJ spheres. The type of a particle is determined by the diameters specified for its 3 shape paramters. If all 3 shape parameters = 0.0, then the particle is treated as an LJ sphere. The epsilon_i_* or epsilon_j_* parameters are ignored for LJ spheres. If the 3 shape paraemters are > 0.0, then the particle is treated as an ellipsoid (even if the 3 parameters are equal to each other).
A12 specifies the energy prefactor which depends on the types of the two interacting particles.
For ellipsoid/ellipsoid interactions, the interaction is computed by the formulas in the supplementary docuement referenced above. A12 is the Hamaker constant as described in (Everaers). In LJ units:
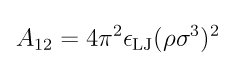
where rho gives the number density of the spherical particles composing the ellipsoids and epsilon_LJ determines the interaction strength of the spherical particles.
For ellipsoid/LJ sphere interactions, the interaction is also computed by the formulas in the supplementary docuement referenced above. A12 has a modifed form (see here for details):
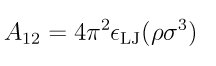
For ellipsoid/LJ sphere interactions, a correction to the distance- of-closest approach equation has been implemented to reduce the error from two particles of disparate sizes; see this supplementary document.
For LJ sphere/LJ sphere interactions, the interaction is computed using the standard Lennard-Jones formula, which is much cheaper to compute than the ellipsoidal formulas. A12 is used as epsilon in the standard LJ formula:
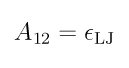
and the specified sigma is used as the sigma in the standard LJ formula.
When one of both of the interacting particles are ellipsoids, then sigma specifies the diameter of the continuous distribution of constituent particles within each ellipsoid used to model the RE-squared potential. Note that this is a different meaning for sigma than the pair_style gayberne potential uses.
The epsilon_i and epsilon_j coefficients are defined for atom types, not for pairs of atom types. Thus, in a series of pair_coeff commands, they only need to be specified once for each atom type.
Specifically, if any of epsilon_i_a, epsilon_i_b, epsilon_i_c are non-zero, the three values are assigned to atom type I. If all the epsilon_i values are zero, they are ignored. If any of epsilon_j_a, epsilon_j_b, epsilon_j_c are non-zero, the three values are assigned to atom type J. If all three epsilon_i values are zero, they are ignored. Thus the typical way to define the epsilon_i and epsilon_j coefficients is to list their values in “pair_coeff I J” commands when I = J, but set them to 0.0 when I != J. If you do list them when I != J, you should insure they are consistent with their values in other pair_coeff commands.
Note that if this potential is being used as a sub-style of pair_style hybrid, and there is no “pair_coeff I I” setting made for RE-squared for a particular type I (because I-I interactions are computed by another hybrid pair potential), then you still need to insure the epsilon a,b,c coefficients are assigned to that type in a “pair_coeff I J” command.
For large uniform molecules it has been shown that the epsilon_*_* energy parameters are approximately representable in terms of local contact curvatures (Everaers):
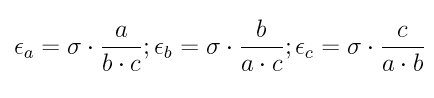
where a, b, and c give the particle diameters.
The last coefficient is optional. If not specified, the global cutoff specified in the pair_style command is used.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section 5 of the manual. The accelerated styles take the same arguments and should produce the same results, except for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages, respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
For atom type pairs I,J and I != J, the epsilon and sigma coefficients and cutoff distance can be mixed, but only for sphere pairs. The default mix value is geometric. See the “pair_modify” command for details. Other type pairs cannot be mixed, due to the different meanings of the energy prefactors used to calculate the interactions and the implicit dependence of the ellipsoid-sphere interaction on the equation for the Hamaker constant presented here. Mixing of sigma and epsilon followed by calculation of the energy prefactors using the equations above is recommended.
This pair styles supports the pair_modify shift option for the energy of the Lennard-Jones portion of the pair interaction, but only for sphere-sphere interactions. There is no shifting performed for ellipsoidal interactions due to the anisotropic dependence of the interaction.
The pair_modify table option is not relevant for this pair style.
This pair style does not support the pair_modify tail option for adding long-range tail corrections to energy and pressure.
This pair style writes its information to binary restart files, so pair_style and pair_coeff commands do not need to be specified in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the inner, middle, outer keywords of the run_style command.
Restrictions
This style is part of the ASPHERE package. It is only enabled if LAMMPS was built with that package. See the Making LAMMPS section for more info.
This pair style requires that atoms be ellipsoids as defined by the atom_style ellipsoid command.
Particles acted on by the potential can be finite-size aspherical or spherical particles, or point particles. Spherical particles have all 3 of their shape parameters equal to each other. Point particles have all 3 of their shape parameters equal to 0.0.
The distance-of-closest-approach approximation used by LAMMPS becomes less accurate when high-aspect ratio ellipsoids are used.