pair_style edip command
Syntax
pair_style edip
pair_style edip/omp
Examples
pair_style edip pair_coeff * * Si.edip Si
Description
The edip style computes a 3-body EDIP potential which is popular for modeling silicon materials where it can have advantages over other models such as the Stillinger-Weber or Tersoff potentials. In EDIP, the energy E of a system of atoms is
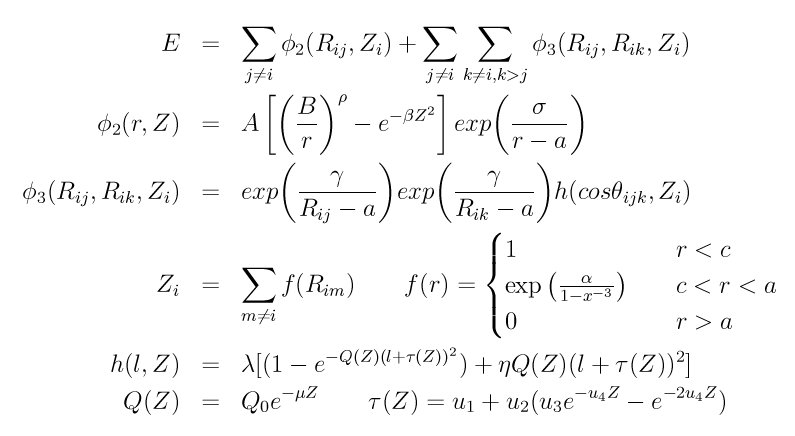
where phi2 is a two-body term and phi3 is a three-body term. The summations in the formula are over all neighbors J and K of atom I within a cutoff distance = a. Both terms depend on the local environment of atom I through its effective coordination number defined by Z, which is unity for a cutoff distance < c and gently goes to 0 at distance = a.
Only a single pair_coeff command is used with the edip style which specifies a EDIP potential file with parameters for all needed elements. These are mapped to LAMMPS atom types by specifying N additional arguments after the filename in the pair_coeff command, where N is the number of LAMMPS atom types:
- filename
- N element names = mapping of EDIP elements to atom types
See the pair_coeff doc page for alternate ways to specify the path for the potential file.
As an example, imagine a file Si.edip has EDIP values for Si.
EDIP files in the potentials directory of the LAMMPS distribution have a ”.edip” suffix. Lines that are not blank or comments (starting with #) define parameters for a triplet of elements. The parameters in a single entry correspond to the two-body and three-body coefficients in the formula above:
- element 1 (the center atom in a 3-body interaction)
- element 2
- element 3
- A (energy units)
- B (distance units)
- cutoffA (distance units)
- cutoffC (distance units)
- alpha
- beta
- eta
- gamma (distance units)
- lambda (energy units)
- mu
- tho
- sigma (distance units)
- Q0
- u1
- u2
- u3
- u4
The A, B, beta, sigma parameters are used only for two-body interactions. The eta, gamma, lambda, mu, Q0 and all u1 to u4 parameters are used only for three-body interactions. The alpha and cutoffC parameters are used for the coordination environment function only.
The EDIP potential file must contain entries for all the elements listed in the pair_coeff command. It can also contain entries for additional elements not being used in a particular simulation; LAMMPS ignores those entries.
For a single-element simulation, only a single entry is required (e.g. SiSiSi). For a two-element simulation, the file must contain 8 entries (for SiSiSi, SiSiC, SiCSi, SiCC, CSiSi, CSiC, CCSi, CCC), that specify EDIP parameters for all permutations of the two elements interacting in three-body configurations. Thus for 3 elements, 27 entries would be required, etc.
At the moment, only a single element parametrization is implemented. However, the author is not aware of other multi-element EDIP parametrizations. If you know any and you are interest in that, please contact the author of the EDIP package.
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the suffix. They have been optimized to run faster, depending on your available hardware, as discussed in Section 5 of the manual. The accelerated styles take the same arguments and should produce the same results, except for round-off and precision issues.
These accelerated styles are part of the GPU, USER-INTEL, KOKKOS, USER-OMP and OPT packages, respectively. They are only enabled if LAMMPS was built with those packages. See the Making LAMMPS section for more info.
You can specify the accelerated styles explicitly in your input script by including their suffix, or you can use the -suffix command-line switch when you invoke LAMMPS, or you can use the suffix command in your input script.
See Section 5 of the manual for more instructions on how to use the accelerated styles effectively.
Mixing, shift, table, tail correction, restart, rRESPA info:
This pair style does not support the pair_modify shift, table, and tail options.
This pair style does not write its information to binary restart files, since it is stored in potential files. Thus, you need to re-specify the pair_style and pair_coeff commands in an input script that reads a restart file.
This pair style can only be used via the pair keyword of the run_style respa command. It does not support the inner, middle, outer keywords.
Restrictions
This angle style can only be used if LAMMPS was built with the USER-MISC package. See the Making LAMMPS section for more info on packages.
This pair style requires the newton setting to be “on” for pair interactions.
The EDIP potential files provided with LAMMPS (see the potentials directory) are parameterized for metal units. You can use the SW potential with any LAMMPS units, but you would need to create your own EDIP potential file with coefficients listed in the appropriate units if your simulation doesn’t use “metal” units.